纤维连接蛋白测定试剂盒(免疫比浊法)产品技术要求jiuqiang
迈瑞医疗 D-二聚体(D-Dimer)测定试剂盒(免疫比浊法)

迈瑞医疗 D-二聚体(D-Dimer)测定试剂盒(免疫比浊法)概述迈瑞医疗 D-二聚体(D-Dimer)测定试剂盒是一种应用于临床实验室的免疫比浊法试剂盒。
D-二聚体是一种特定的血液标志物,其浓度的升高通常与凝血系统活化或纤维蛋白溶解活动增加相关。
迈瑞医疗 D-二聚体测定试剂盒可用于定性和定量测定体液中的D-二聚体水平,以辅助临床医生对深静脉血栓(DVT)和肺栓塞(PE)等血栓栓塞性疾病的诊断和监测。
技术原理迈瑞医疗 D-二聚体测定试剂盒采用免疫比浊法进行测定。
试剂盒中包含特异性抗体对D-二聚体进行识别和结合。
首先,样本中的D-二聚体与与其特异性结合的抗体反应形成免疫复合物。
然后,将金标记的二抗加入反应体系中,与免疫复合物结合,并形成比浊体系。
比浊体系中的免疫复合物会散射光线,导致测光仪器测量到的吸光度增加。
测光仪器会根据吸光度的变化,计算出样本中D-二聚体的浓度。
试剂盒组成迈瑞医疗 D-二聚体测定试剂盒包含以下组分:1.D-二聚体抗体:特异性地与D-二聚体结合的抗体。
2.金标记的二抗:与免疫复合物结合,并形成比浊体系。
3.样本稀释液:用于稀释样本,以确保在免疫反应中得到准确的结果。
4.试剂盘:包含预涂有特异性抗体的孔板。
操作步骤以下是使用迈瑞医疗 D-二聚体测定试剂盒进行分析的基本操作步骤:1.取出试剂盘,标注每个孔的编号。
2.加入待测样本和对照品到相应的孔中。
3.使用分注器向每个孔中加入适量的稀释液。
4.轻轻摇动试剂盘,使试剂充分混合。
5.将试剂盘放入测光仪器中,按照仪器的操作说明进行测量。
6.根据测量结果,参照试剂盒说明书,计算样本中D-二聚体的浓度。
结果解读迈瑞医疗 D-二聚体测定试剂盒的结果解读应结合临床病史和其他诊断方法进行综合分析。
一般来说,D-二聚体浓度的升高可能与以下情况相关:•深静脉血栓(DVT)•肺栓塞(PE)•动脉栓塞•心肌梗死•外科手术或创伤后•某些肿瘤恶化然而,D-二聚体浓度的升高并不能直接说明某种特定疾病的存在,因此,医生应该结合临床病史和其他检查结果来做出准确的诊断。
产品技术规格偏离表
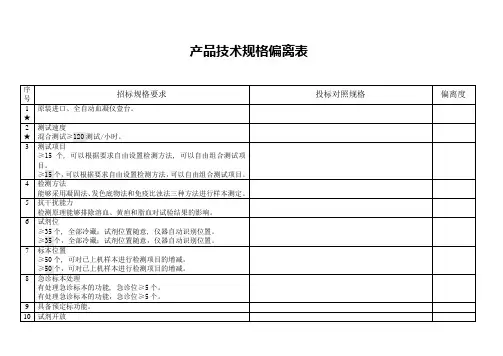
网络连接
实验结果直接进入LIS系统, 通过HIS向临床科室直接发送报告。具有网络接口, 数据输出接口。
实验结果直接进入LIS系统,通过HIS向临床科室直接发送报告。具有网络接口,数据输出接口。
13
配盖帽穿刺功能及ID条码阅读器。
14
触摸式液晶显示屏。
15
备件、专用工具、资料及其它
①卖方应随机向买方提供一套标准件包, 并列出清单及单价。
投标单位签字(盖章):
④保修三年,终身维护。提供24小时维修联络方法,接到报修后4小时到位,假如仪器出现故障,一周内未能修复,应提供备用机,以供用户应急使用,软件免费升级。
17
技术培训要求
①卖方应提供现场技术培训,保证使用人员正常操作设备的各种功能。
②根据设备要求, 应向买方提供使用和维修技术人员培训。
②根据设备要求,应向买方提供使用和维修技术人员培训。
②为保证设备正常运行, 卖方应在中国境内设置备件库, 存入所有必须的备件, 保证10年以上的供应期, 并出具证明文件。
③卖方必须向买方提供操作手册一套。
④卖方须向买方提供设备的运行、安装、使用环境要求以及设备维护的专用工具。
⑤提供凝血常规项目成本核算清单和临床检验收费许可文件。
16
★
技术服务
①在货物到达使用单位后, 卖方应在7天内派工程技术人员到达现场, 在买方技术人员在场的情况下开箱清点货物, 组织安装、调试、并承担因此发生的一切费用。
5
抗干扰能力
检测原理能够排除溶血、黄疸和脂血对试验结果的影响。
6
试剂位
≥35个, 全部冷藏;试剂位置随意, 仪器自动识别位置。
≥35个,全部冷藏;试剂位置随意,仪器自动识别位置。
类体外诊断试剂《产品技术要求》.docx

医疗器械产品技术要求编号:*****检测试剂盒(胶体金法)1.产品型号 / 规格及划分说明试剂盒规格为 10 人份 / 盒, 每个试剂盒内装10 包检测试剂、 1 瓶抽提液 A、1 瓶抽提液 B、10 个采样管、 10 个小滴管和 1 份说明书。
2.性能指标物理检查外观包装盒整洁,各组分齐全;抽提液瓶盖严紧,无漏液;盒签和瓶签清晰完整,标识无误;品名、批号和有效期清楚;说明书清晰完整。
膜条宽度膜条宽度±。
液体移行速度液体移行速度应不低于10mm/min。
装量抽提液装量为±瓶。
阳性参考品符合率10 份*** 国家阳性参考品检测结果应全部为阳性,阳性参考品符合率(+/+ )应为 10/10 或 3 份企业阳性参考品检测结果应全部为阳性,阳性参考品符合率(+/+ )应为 3/3 。
阴性参考品符合率10 份*** 国家阴性参考品检测结果应全部为阴性,阴性参考品符合率(-/- )应为 10/10 或 10 份企业阴性参考品检测结果应全部为阴性,阴性参考品符合率(-/- )应为 10/10 。
重复性使用 *** 国家重复性参考品或企业重复性参考品进行检测,结果应为阳性,且显色一致无明显差别。
最低检出限使用 *** 国家最低检出限参考品或企业最低检出限参考品进行检测,结果应为阳性,最低检出限应不高于 1×105个菌 /ml 。
批间差使用 *** 国家重复性参考品或企业重复性参考品对三批试剂盒进行检测,结果应符合的规定。
稳定性试剂盒在规定的存储条件下存放至有效期末或37℃条件下放置14 天,分别检测 ~项,结果应符合各项目的要求。
3.检验方法物理检查外观在自然光下目视检查,结果应符合的要求。
膜条宽度随机抽取 5 支试剂条,用游标卡尺测量宽度,结果应符合的要求。
液体移行速度随机抽取 5 支试剂条,将试剂条平放,在试剂条加样孔中滴加 3 滴抽提液,开始用秒表计时,直到液体完全通过试剂条区域时停止计时,所用时间记为(t ),用游标卡尺测量从加样孔至试剂条末端的长度,记为(L),计算 L/t 即为移行速度,结果应符合的要求。
转铁蛋白检测试剂盒(免疫比浊法)说明书

转铁蛋白检测试剂盒(免疫比浊法)说明书转铁蛋白检测试剂盒(免疫比浊法)说明书【产品名称】通用名称:转铁蛋白检测试剂盒(免疫比浊法)英文名称:TRF Kit【包装规格】R1:R2 ,1×30ml;1×10ml1×60ml;1×20ml 2×60ml;2×20ml【预期用途】转铁蛋白检测试剂盒临床上用于定量测定人体血清中转铁蛋白的含量。
转铁蛋白(TRF)连接上铁离子之后可以防止铁中毒以及通过肾的流失。
其水平的升高常见于铁缺乏症、怀孕、雌性激素的控制以及类脂肪的肾病。
其水平的降低常见于睾丸激素的控制、感染、急性的炎症、某些类型的肾炎、血色素缺失、急性的疟疾以及营养不良。
【检验原理】人体中的转铁蛋白与试剂中抗人转铁蛋白抗体在缓冲液中快速形成抗原抗体复合物,使反应液出现浊度。
当反应液中保持抗体过剩时,形成的复合物随抗原量增加而增加,反应液的浊度亦随之增加,在340nm以终点法检测吸光度变化,与校准品对照,即可计算出未知蛋白的含量。
【主要组成成分】组成主要成分R1 NaH2PO4缓冲液R2 抗人转铁蛋白抗体注意不同批号的试剂盒的组分不能混用。
校准品:用户自行购买利德曼公司的多项高值免疫标准液,标准值见说明书;质控品:用户自行购买利德曼公司多项免疫质控血清,质控值见说明书;【储存条件及有效期】1.包装试剂均应在2?,8?避光储存,可稳定至标签所示失效日期;2( 试剂有效期为12个月;3( 开瓶有效期:10天(开瓶后在2?,8?保存);【适用仪器】包装规格适用机型1×30ml;1×10ml 日立7060、1×60ml;1×20ml 日立7170、东芝-40 2×60ml;2×20ml 日立7020、奥林巴斯AU640、贝克曼CX4 【样本要求】1、标本为离心或分离除去血液凝块的新鲜血清。
2、血清样本在2~8?储存不超过一周。
全自动凝血分析仪产品技术要求九强

全自动凝血分析仪组成:产品由光学检测模块、温控模块、机械臂模块、反应杯进给模块、液路模块、主控模块构成。
适用范围:本仪器用于对血液进行凝血和抗凝、纤溶和抗纤溶等功能的分析。
通过采用凝固法、发色底物法和免疫比浊法,与配套的检测试剂共同使用,供临床对血液凝血酶原时间(PT)、活化部分凝血活酶时间(APTT)、凝血酶时间(TT)、纤维蛋白原(FIB)、D-二聚体(DD)、纤维蛋白(原)降解产物(FDP)、抗凝血酶Ⅲ(ATⅢ)进行定量分析。
2.1.预温时间仪器开机后,应在30分钟内达到仪器的工作温度,即37.0℃±1.0℃。
2.2 温度控制2.2.1 仪器开机稳定后,检测和温育位恒温装置部的反应体系温度控制在37.0℃±1.0℃范围内。
2.2.2 仪器开机稳定后,试剂冷却位温度控制应不超过16.0℃。
2.3 检测项目和报告单位检测项目包括血浆凝血酶原时间(PT)、活化部分凝血活酶时间(APTT)、纤维蛋白原(FIB)、纤维蛋白(原)降解产物(FDP)、凝血酶时间(TT)、D-二聚体(DD)和ATⅢ的测定。
PT、APTT、TT的报告单位为秒(s),其中PT的测定结果应报告国际标准化比值(INR);FIB的报告单位为g/L或mg/dL;FDP的报告单位为μg/mL;DD的报告单位为ng/mL;ATⅢ的报告单位为%。
2.4携带污染率2.4.1 样品浓度的携带污染率:FIB、DD和ATⅢ的携带污染率应≤10%。
2.4.2 项目携带污染率:FIB对PT的携带污染率应≤10%。
DD对PT的携带污染率≤10%,抗凝血活酶Ⅲ(ATⅢ)对PT的携带污染率≤10%。
2.5 测试速度仪器的测试速度PT约为360测试/小时;DD每小时不低于120个测试;ATⅢ每小时不低于130个测试。
2.6 精密度分别用正常样本、异常样本的血浆进行测试,分析仪的精密度应符合表1的要求。
表1 不同凝血试验测定项目的精密度要求注:异常样本指不小于仪器正常参考范围中位值两倍值。
关于电子宫腔观察镜61个产品分类界定的通知

关于电子宫腔观察镜61个产品分类界定的通知各省、自治区、直辖市食品药品监督管理局:为适应医疗器械监督管理工作的需要,总局组织有关单位和专家对腹腔镜手术用内窥镜自动调控定位装置等61个产品的管理类别进行了界定。
现通知如下:一、作为Ⅲ类医疗器械管理的产品(5个)(一)腹腔镜手术用内窥镜自动调控定位装置:主要由主机、聚焦模块、头盖(信号发射用)、脚踏板、显示器组成。
通过信号发射头盖和显示器来决定硬镜的活动方向和位置,然后踩脚踏板可将硬镜活动到所需位置和方向,与患者接触。
用于在腹腔镜术中协助医生操作硬镜的位置和方向。
分类编码:6822。
(二)髓内钉延长系统:由磁手柄、控制面板及电源适配器组成。
与特定的可延长髓内钉配合使用,用于控制髓内钉在髓腔里的缩回或伸长。
分类编码:6821。
(三)电子交叉配血系统:通过采集献血者血袋及患者的血型和抗体筛查检测结果,对采集数据进行确认,在抗体筛查结果均为阴性的条件下,匹配相同血型的献血者与患者血液。
可以独立使用,也可与血液配型设备配合使用。
用于指导医疗机构输血前配血工作。
分类编码:6870。
(四)数字化X射线透视摄影系统:由高压发生器、X射线管组件、限束器、诊断床、图像处理装置、平板探测器组成。
用于对患者的头部、躯干、四肢进行数字X射线摄影、透视和胃肠诊断、数字减影血管造影、体层摄影、泌尿摄影。
分类编码:6830。
(五)电子宫腔观察镜:由镜芯(包含CMOS摄像头和LED冷光源)、操作手柄、数据传输线等组成。
与一次性镜鞘配套使用,通过阴道进入子宫宫腔进行观察。
用于辅助医生观察宫腔内情况。
分类编码:6822。
二、作为Ⅱ类医疗器械管理的产品(30个)(一)癫痫发作报警器:由床垫传感器和电路盒组成。
床垫传感器可以检测人体的动作信息,从而转化为电信号。
用于癫痫患者或小儿惊厥患者睡眠状态下,癫痫发作时即时报警。
分类编码:6821。
(二)睑板腺热脉动治疗仪:由主机以及一次性使用无菌眼睑治疗头(包含眼杯和眼睑加热器等)组成。
纤维蛋白(原)降解产物测定试剂盒(胶乳免疫比浊法)产品技术要求赛科希德
纤维蛋白(原)降解产物测定试剂盒(胶乳免疫比浊法)适用范围:本产品用于体外定量测定人血浆中纤维蛋白(原)降解产物(FDP)含量。
1.1 包装规格见表1。
表1 包装规格1.2 主要组成成分本产品由FDP试剂1、FDP试剂2组成。
FDP试剂1主要成分为2%三羟甲基氨基甲烷缓冲液、1‰苯甲酸钠;FDP试剂2主要成分为0.25%鼠抗人FDP单克隆抗体胶乳颗粒、0.5%牛血清白蛋白。
2.1 外观2.1.1 试剂盒包装完整,标签应清晰易识别。
2.1.2 FDP试剂1应为无色均匀液体,FDP试剂2应为白色均匀乳液。
2.2 空白吸光度试剂空白吸光度差值(△A)≤0.02。
2.3 灵敏度FDP 10μg/mL时,吸光度差值(△A)应≥0.100。
2.4 线性范围2.4.1在[2.5,40]μg/mL线性范围内,相关系数γ≥0.98。
2.4.2线性偏差:[2.5,5]μg/mL时绝对偏差不超过±0.5μg/mL,(5,40]μg/mL 时相对偏差不超过±10%。
2.5 准确度与已上市产品作比对,取40个[2.5,40]μg/mL浓度范围内不同浓度的人源样本,计算相关系数应≥0.975。
[2.5,5]μg/mL的样本绝对偏差不超过±0.5μg/mL,(5,40]μg/mL的样本相对偏差不超过±10%。
2.6 重复性用同一批号FDP试剂盒重复水平Ⅰ、水平Ⅱ两个水平的质控品,所得结果变异系数(CV)≤10%。
2.7 批间差批间极差≤8%。
2.8 装量FDP试剂盒中液体性状试剂的装量不少于其试剂瓶瓶签的标称量。
2.9 稳定性FDP试剂盒在2℃~8℃密闭的贮存条件下保存至有效期12个月后3个月内,产品的性能应符合2.1、2.2、2.3、2.4、2.5、2.6条规定的要求。
国家食品药品监督管理总局公告 第号
国家食品药品监督管理总局公告2017年第38号关于批准发布《口腔医疗器械生物学评价第7部分:牙髓牙本质应用试验》等40项医疗器械行业标准的公告(2017年第38号)YY/T0127.7-2017《口腔医疗器械生物学评价第7部分:牙髓牙本质应用试验》等40项医疗器械行业标准已经审定通过,现予以公布。
标准自2018年4月1日起实施,标准编号、名称及适用范围见附件。
特此公告。
附件:YY/T0127.7-2017《口腔医疗器械生物学评价第7部分:牙髓牙本质应用试验》等40项医疗器械行业标准编号、名称及适用范围食品药品监管总局2017年3月28日序号标准编号标准名称代替标准号发布日期实施日期1YY/T0127.7-2017口腔医疗器械生物学评价第7部分:牙髓牙本质应用试验YY/T0127.7-20012017-03-282018-04-012YY/T0492-2017植入式心脏起搏器电极导线YY/T0492-20042017-03-282018-04-013YY/T0653-2017血液分析仪YY/T0653-20082017-03-284YY/T0654-2017全自动生化分析仪YY/T0654-20082017-03-282018-04-015YY/T0657-2017医用离心机YY/T0657-20082017-03-282018-04-016YY/T0659-2017凝血分析仪YY/T0658-2008 YY/T0659-20082017-03-282018-04-017YY/T0686-2017医用镊YY/T0686-20082017-03-282018-04-018YY/T0706-2017乳腺X射线机专用技术条件YY/T0706-20082017-03-282018-04-019YY/T1122-2017咬骨钳(剪)通用技术条件YY1122-2005YY/T1127-20062017-03-282018-04-0110YY/T1403-2017环氧乙烷分包灭菌的要求2017-03-282018-04-0111YY/T1465.4-2017医疗器械免疫原性评价方法第4部分:小鼠腹腔巨噬细胞吞噬鸡红细胞试验半体内法2017-03-282018-04-0112YY/T1510-2017医用血浆病毒灭活箱2017-03-282018-04-01C反应蛋白测定试剂盒2017-03-282018-04-0114YY/T1516-2017泌乳素定量标记免疫分析试剂盒2017-03-282018-04-0115YY/T1518-2017C-肽(C-P)定量标记免疫分析试剂盒2017-03-282018-04-01电生理标测导管2017-03-282018-04-0117YY/T1521-2017超声弹性仿组织体模的技术要求2017-03-282018-04-0118YY/T1522-2017连接到医用气体管道系统终端的流量测量装置2017-03-282018-04-01二氧化碳测定试剂盒(PEPC酶法)2017-03-282018-04-0120YY/T1524-2017α-L-岩藻糖苷酶(AFU)测定试剂盒(CNPF底物法)2017-03-282018-04-0121YY/T1527-2017α/β-地中海贫血基因分型检测试剂盒2017-03-282018-04-01YY/T1528-2017肌红蛋白测定试剂盒(免疫比浊法)2017-03-282018-04-0123YY/T1530-2017尿液有形成分分析仪用控制物质2017-03-282018-04-0124YY/T1531-2017细菌生化鉴定系统2017-03-282018-04-01YY/T1532-2017医疗器械生物学评价纳米材料溶血试验2017-03-282018-04-0126YY/T1533-2017全自动时间分辨荧光免疫分析仪2017-03-282018-04-0127YY/T1534-2017医用LED设备光辐射安全分类的检测方法2017-03-282018-04-0128YY/T1535-2017人类体外辅助生殖技术用医疗器械生物学评价人精子存活试验2017-03-282018-04-0129YY/T1536-2017非血管内导管表面滑动性能评价用标准试验模型2017-03-282018-04-0130YY/T1555.1-2017硅凝胶填充乳房植入物专用要求硅凝胶填充物性能要求第1部分:易挥发性物质限量要求2017-03-2831YY/T1556-2017医用输液、输血、注射器具微粒污染检验方法2017-03-282018-04-0132YY/T1557-2017医用输液、输血、注射器具用热塑性聚氨酯专用料2017-03-282018-04-0133YY/T1559-2017脊柱植入物椎间盘假体静态及动态性能试验方法2018-04-0134YY/T1560-2017脊柱植入物椎体切除模型中枕颈和枕颈胸植入物试验方法2017-03-282018-04-0135YY/T1561-2017组织工程医疗器械产品动物源性支架材料残留α-Gal抗原检测测2017-03-282018-04-0136YY/T1562-2017组织工程医疗器械产品生物材料支架细胞活性试验指南2017-03-282018-04-0137YY/T1563-2017脊柱植入物全椎间盘假体功能、运动和磨损评价试验方法2017-03-282018-04-0138YY/T1564-2017心血管植入物肺动脉带瓣管道体外脉动流性能测试方法2017-03-282018-04-0139YY/T1565-2017外科植入物无损检验铸造金属外科植入物射线照相检验2017-03-282018-04-0140YY/T1568-2017子宫托2017-03-282018-04-01。
人纤维蛋白原(Fbg)酶联免疫吸附测定试剂盒说明书
人纤维蛋白原(Fbg)酶联免疫吸附测定试剂盒使用说明书本试剂盒仅供体外研究使用、不用于临床诊断!预期应用ELISA法定量测定人血清、血浆或其它相关生物液体中Fbg含量。
实验原理用纯化的Fbg抗体包被微孔板,制成固相载体,往微孔中依次加入标本或标准品、生物素化的Fbg抗体、HRP标记的亲和素,经过彻底洗涤后用底物(TMB)显色。
TMB在过氧化物酶的催化下转化成蓝色,并在酸的作用下转化成最终的黄色。
颜色的深浅和样品中的Fbg呈正相关。
用酶标仪在450nm波长下测定吸光度(值),计算样品浓度。
试剂盒组成及试剂配制1、酶标板:一块(96孔)2、标准品(冻干品):2瓶,请临用前15分钟内配制。
每瓶以样品稀释液稀释至1ml,盖好后室温静置大约10分钟,同时反复颠倒/搓动以助溶解,其浓度为2,500ng/ml,然后做系列倍比稀释(注:不要直接在板中进行倍比稀释),分别配制成2,500ng/ml,1,250ng/ml,625ng/ml,312ng/ml,156ng/ml,78ng/ml,39 ng/ml,样品稀释液直接作为空白孔0ng/ml。
如配制1,250ng/ml标准品:取0.5ml(不要少于0.5ml)2,500 ng/ml的上述标准品加入含有0.5ml样品稀释液的Eppendorf管中,混匀即可,其余浓度以此类推。
3、样品稀释液:1×20ml。
4、检测稀释液A:1×10ml。
5、检测稀释液B:1×10ml。
6、检测溶液A:1×120/瓶(1:100)。
临用前以检测稀释液A1:100稀释(如:10检测溶液A/990检测稀释液A),充分混匀,稀释前根据预先计算好的每次实验所需的总量配制(100/孔),实际配制时应多配制0.1-0.2ml。
7、检测溶液B:1×120/瓶(1:100)。
临用前以检测稀释液B1:100稀释。
稀释方法同检测溶液A。
8、底物溶液:1×10ml/瓶。
特定蛋白分析仪产品技术要求sj
特定蛋白分析仪适用范围:已上市的免疫比浊试剂配套使用,用于临床机构测量人体血清中特定蛋白的浓度。
1.1特定蛋白免疫分析仪产品型号划分说明:1.2产品组成:分析仪主要由控制系统(包括显示、键盘)、光学系统、打印系统、温控系统和电源适配器组成。
1.3各型号间差异说明:工作条件环境温度:15℃~35℃;相对湿度:≤95%;大气压力:860hPa~1060hPa;电源:A.C.220V、50Hz;预热:≥10min;仪器附近无强磁场干扰,无剧烈震动,无强光直射。
1.1 外观:外观应满足以下要求:1.1.1 面板上的图形符号和文字应准确、清晰、均匀、不得有划痕;1.1.2 紧固件连接应牢固可靠,不得有松动;1.1.3 运动部件应平稳,不应卡住、突跳及显著空回,摩擦阻力均匀,键组回跳应灵活。
2.2 杂散光:测量无浊度水时,浊度≤1(FTU)。
2.3 线性范围:分析仪测量浊度在10-300(FTU)的浊度液时,线性偏倚不大于±6%,线性相关系数不小于0.975。
2.4 浊度准确度:应符合表1的规定。
分析仪测量浊度准确性符合表1的规定。
表 1 浊度准确性2.5 散射光强重复性:散射光强重复性CV≤4%。
2.6 浊度的稳定性:分析仪在开机30min后,4hr内测量一次浊度为200FTU的福马肼浊度液、在第5hr再测量一次浊度为200FTU的福马肼浊度液,其变化的相对极差不大于3%。
2.7 测量通道间的一致性(OET-N400适用)OET-N400有4个独立的测量通道,在测量浊度为150FTU的福马肼浊度液时,各通道之间测量值极差不大于4FTU。
2.8 测量孔温度准确性与温度波动:分析仪测量孔的恒温温度为37℃,其实测恒温温度准确性误差小于±0.5℃,实测温度波动不大于±0.2℃。
2.9 临床项目的批内精密度:变异系数(CV)应满足表2的要求:表2 临床项目的批内精密度要求2.10 电气安全产品属于过压类别II类,污染等级2级,应符合GB 4793.1-2007、GB 4793.6-2008、GB 4793.9-2013及YY 0648-2008的要求,产品安全特征见附录A。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
纤维连接蛋白测定试剂盒(免疫比浊法)
适用范围:用于体外定量测定人血清或血浆中纤维连接蛋白的含量。
1.1 包装规格
包装规格见表1。
表1 包装规格
1.2 主要组成成分
主要组成成分见表2。
表2主要组成成分
注:不同批号的校准品、质控品赋值有差异,具体赋值详见靶值单。
2.1 外观
试剂1为无色澄清液体,目测不得有任何沉淀及絮状悬浮物;
试剂2为无色澄清液体,目测不得有任何沉淀及絮状悬浮物;
校准品为黄色澄清液体,目测不得有任何沉淀及絮状悬浮物;
质控品为黄色澄清液体,目测不得有任何沉淀及絮状悬浮物。
试剂盒标签标识清晰,外包装完整无损。
2.2 净含量
试剂的净含量应不少于标称量。
2.3 试剂空白吸光度
A340nm下测定空白吸光度应≤ 0.5000。
2.4 准确度
与已上市产品进行比对试验:相关系数r≥0.975,在[20.0,100.0] mg/L区间内测定的绝对偏差应不超过±10 mg/L,在(100.0,600.0] mg/L区间内测定的相对偏差应不超过±10%。
2.5 分析灵敏度
样本浓度为150 mg/L时,其吸光度变化在0.0200~0.1500之间。
2.6 线性区间
在[20.0,600.0] mg/L区间内,线性相关系数r≥0.990,在[20.0,100.0] mg/L 区间内测定的绝对偏差应不超过±10 mg/L,在(100.0,600.0] mg/L区间内测定的相对偏差应不超过±10%。
2.7 测量精密度
2.7.1 重复性
对高、低不同浓度的同一血清样本或质控品重复测定10次,其测定值的变异系数(CV%)应不大于10%。
2.7.2 批间差
随机抽取三批试剂盒的批间相对极差(R)应不大于10%。
2.8 稳定性
试剂盒在2℃~8℃密封避光保存,有效期为12个月。
在试剂盒有效期满后一个月以内,应符合2.1、2.3、2.4、2.5、2.6、2.7.1的要求。
2.9校准品溯源性
按照《GB/T 21415-2008体外诊断医疗器械生物样品中量的测量校准品和控制物质赋值的计量学溯源性》的要求,提供所有产品校准的来源、赋值过程以及测量不确定度,试剂盒校准品溯源至上海北加公司的纤维结合蛋白测定试剂盒。