蛋白质分析路线、方法
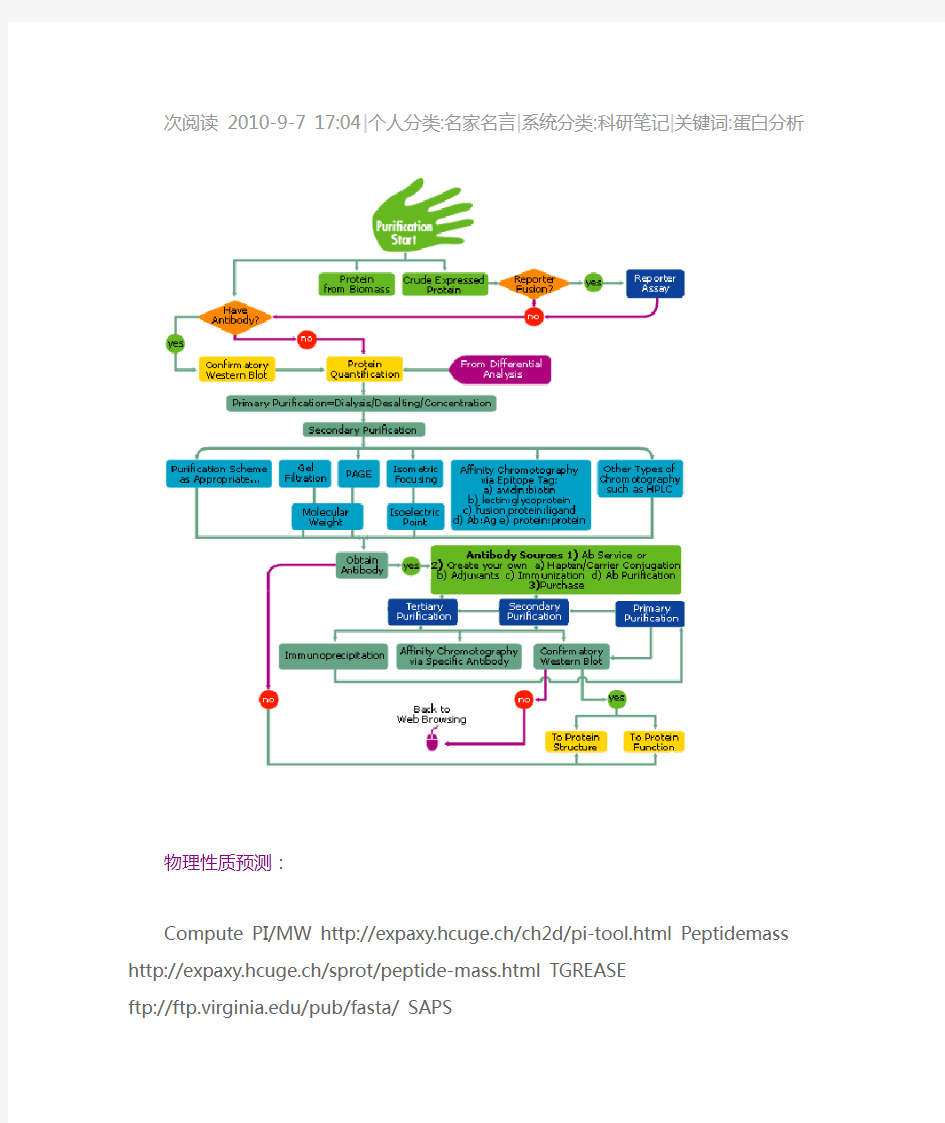

次阅读2010-9-7 17:04|个人分类:名家名言|系统分类:科研笔记|关键词:蛋白分析
物理性质预测:
Compute PI/MW http://expaxy.hcuge.ch/ch2d/pi-tool.html Peptidemass
http://expaxy.hcuge.ch/sprot/peptide-mass.html TGREASE
ftp://https://www.360docs.net/doc/929655676.html,/pub/fasta/ SAPS http://ulrec3.unil.ch/software/SAPS_form.html
基于组成的蛋白质识别预测
AACompIdent http://expaxy.hcuge.ch/ch2d/aacompi.html AACompSim
http://expaxy.hcuge.ch/ch2d/aacsim.html PROPSEARCH
http://www.embl-heidelberg.de/prs.html
二级结构和折叠类预测
nnpredict https://www.360docs.net/doc/929655676.html,/~nomi/nnpredictPredictprotein
http://www.embl-heidelberg.de/predictprotein/SOPMA
http://www.ibcp.fr/predict.htmlSSPRED
http://www.embl-heidelberg.de/sspred/ssprd_info.html
特殊结构或结构预测
COILS http://ulrec3.unil.ch/software/COILS_form.htmlMacStripe
https://www.360docs.net/doc/929655676.html,/matsudaira/macstripe.html
与核酸序列一样,蛋白质序列的检索往往是进行相关分析的第一步,由于数据库和网络技校术的发展,蛋白序列的检索是十分方便,将蛋白质序列数据库下载到本地检索和通过国际互联网进行检索均是可行的。
由NCBI检索蛋白质序列
可联网到:“https://www.360docs.net/doc/929655676.html,:80/entrz/query.fcgi?db=protein”进行检索。
利用SRS系统从EMBL检索蛋白质序列
联网到:https://www.360docs.net/doc/929655676.html,/”,可利用EMBL的SRS系统进行蛋白质序列的检索。
通过EMAIL进行序列检索
当网络不是很畅通时或并不急于得到较多数量的蛋白质序列时,可采用EMAIL方式进行序列检索。
蛋白质基本性质分析
蛋白质序列的基本性质分析是蛋白质序列分析的基本方面,一般包括蛋白质的氨基酸组成,分子质量,等电点,亲水性,和疏水性、信号肽,跨膜区及结构功能域的分析等到。蛋白质的很多功能特征可直接由分析其序列而获得。例如,疏水性图谱可通知来预测跨膜螺旋。同时,也有很多短片段被细胞用来将目的蛋白质向特定细胞器进行转移的靶标(其中最典型的例子是在羧基端含有KDEL序列特征的蛋白质将被引向内质网。WEB中有很多此类资源用于帮助预测蛋白质的功能。
疏水性分析
位于ExPASy的ProtScale程序( https://www.360docs.net/doc/929655676.html,/cgi-bin/protscale.pl)可被用来计算蛋白质的疏水性图谱。该网站充许用户计算蛋白质的50余种不同属性,并为每一种氨基酸输出相应的分值。输入的数据可为蛋白质序列或SWISSPROT数据库的序列接受号。需要调整的只是计算窗口的大小(n)该参数用于估计每种氨基酸残基的平均显示尺度。
进行蛋白质的亲/疏水性分析时,也可用一些windows下的软件如, bioedit,dnamana等。
跨膜区分析
有多种预测跨膜螺旋的方法,最简单的是直接,观察以20个氨基酸为单位的疏水性氨基酸残基的分布区域,但同时还有多种更加复杂的、精确的算法能够预测跨膜螺旋的具体位置和它们的膜向性。这些技术主要是基于对已知跨膜螺旋的研究而得到的。自然存在的跨膜螺旋Tmbase 数据库,可通过匿名FTP获得(http://www.isrec.isb-sib.ch/ftp-server/tmbase),参见表一
资源名称网址说明
TMPRED https://www.360docs.net/doc/929655676.html,/software/TMPRED_form.html 基于对tmpred数据库的统计分析PHDhtm
http://www.embl-heidelberg.de/services/sander/predictprotein/predictprotein.htmlMEM SAT ftp://https://www.360docs.net/doc/929655676.html, 微机版本
,蛋白质序列含有跨膜区提示它可能作为膜受体起作用,也可能是定位于膜的锚定蛋白或者离子通道蛋白等,从而,含有跨膜区的蛋白质往往和细胞的功能状态密切相关。
http://genome.cbs.dtu.dk/sevices/TMHMM-2.0“或
“https://www.360docs.net/doc/929655676.html,/software/TMPRED_form.html”
前导肽与蛋白质定位
在生物内,蛋白质的合成场所与功能场所常被一层或多层细胞膜所隔开,这样就涉及到蛋白质的转运。合成的蛋白质只有准确地定向运行才能保证生命活动的正常进行。一般来说,蛋白质的定位的信息存在于该蛋白质自身结构中,并通过与膜上特殊的受体相互作用而得以表达。在起始密码子之后,有一段编码疏水性氨基酸序列的RN***段,这个氨基酸序列就这个氨基酸序列就是信号肽序列。含有信号肽的蛋白质一般都是分泌到细胞外,可能作为重要的细胞因子起作用,从而具有潜在的应用价值。
http://genome.cbs.dtu.dk/sevices/signalP-2.0
卷曲螺旋分析
另一个能够直接从序列中预测的功能motif是α-螺旋的卷曲排列方式。在这种结构中,两种螺旋通过其疏水性界面相互缠在一起形成一个十分稳定的结构。
蛋白质卷曲的相关资源
资源网址
coiled-coil https://www.360docs.net/doc/929655676.html,/depts/biol/units/coils/coilcoil.htmlCOILS
https://www.360docs.net/doc/929655676.html,/software/COILS_form.htmlEpitopeInfo
https://www.360docs.net/doc/929655676.html,/Links.htm
蛋白质功能预测
基于序列同源性分析的蛋白质功能预测
到少有80个氨基酸长度范围内具有25%以上序列一致性才提示可能的显著性意义。最快的工具
如BlastP能很容易地发现显著性片段,而无需使用十分耗时的BLITZ软件。
基于NCBI/BLAST软件的蛋白序列同源性分析
类似于核酸序列同源性分析,用户直接将待分析的蛋白质序列输入
NCBI/BLAST(https://www.360docs.net/doc/929655676.html,/blast),选择程序BLASTP就可网上分析。
基于WU/BLAST2软件进行分析
华盛顿大学的BLAST软件(dove.embl-heidelberg.dl/blast2)也可进行蛋白质序列的同源性分析。
基于motif、结构位点、结构功能域数据库的蛋白质功能预测
蛋白质的磷酸化与糖基化对蛋白质的功能影响很大,所以对其的分析也是生物信息学的一个部分。同时,分子进化方面的研究表明,蛋白质的不同区域具有不同的进化速率,一些氨基酸必须在进化过程中足够保守以实现蛋白质的功能。在序列模式的鉴定方面有两类技术,第一类是依赖于和一致性序列(consensus sequence)或基序各残基的匹配模式,该技术可用于十分容易并快速搜索motif数据库。
Motif数据库-PROSITE
最好的是PROSITE(https://www.360docs.net/doc/929655676.html,/prosite)
蛋白质序列的(profile)分析
www.isrec.isb-sib.ch/software/PFSCAN_form.html
InterProScan综合分析网站
InterProScan是EBI 开发的一个集成了蛋白质结构域和功能位点的数据库,其中把
SWISS-PROT,TrEMBL.PROTSITE.PRINTS.PFAM.ProDom等数据库提供的蛋白质序列中的各种局域
模式,如结构域,motif等信息统一起来,提供了一个较为全央的分析工具。
https://www.360docs.net/doc/929655676.html,/interpro/scan.html
蛋白质的结构功能域分析
简单模块构架搜索工具(simple modular architecture research tool,SMART)一个较好的蛋白质结构功能域的数据,可用于蛋白质结构功能域的分析,所得到的结构域同时提供相关的资源的链接http://smart.embl-heidelberg.de/
蛋白质结构预测
PDB数据库
蛋白质基本立体结构数据库(PDB, https://www.360docs.net/doc/929655676.html,)其中有大量工具用于查看PDB数据库中的结构,如rasmol,可用于显于出蛋白质的空间结构,下载地址:https://www.360docs.net/doc/929655676.html,/microbio/rasmol)
PDBFinder 数据库
是在PDB、DSSP、HSSP基础上建立的二级库,它包含PDB序列,作者,R因子,分辨率、二级结构等,这些些信息随着PDB库每次发布新版,PDBFinder在EBI自动生成,网址为
“www.sander.embl-heideberg.de/pdbfinder/ ftp://swift.embl-heidelberg.de/pdbfinder.
NRL-3D数据库
是所有已知结构蛋白质的数据库,可用于查询蛋白序列时行相似性分析以确定其结构,
https://www.360docs.net/doc/929655676.html,/Dan/protein/nrl3d.html
ISSD数据库
蛋白质序列数据库,其每个条目包含一个基因的编码序列,同相应的氨基酸序列对比,并给出相应的多肽链结构数据。www.protein.bio.msu.su/issd
HSSP数据库
是根据同源性导出的蛋白质二级结构数据库,每一条PDB项目都有一个对应的HSSP文件,www.sander.embl-heidelberg.de/hssp
蛋白质结构分类数据库
对已知蛋白质三维结构进行手工分类得到的数据库,位于剑桥的站点也提供BLAST检索服务https://www.360docs.net/doc/929655676.html,/scop/
MMDB蛋白质分子模型数据库
是ENTREZ检索工具所使用的三维结构数据库,以ASN格式反蚋的PDB中的结构和序列数据。NCBI 同时提供一个配套的三维结构显示程序的Cn3D,https://www.360docs.net/doc/929655676.html,/Structure/
Dali/FSSP数据库
基于PDB数据库中现有的蛋白质三维结构,用自动结构对比程序Dali比较而形成的折叠单元和家庭分类库。https://www.360docs.net/doc/929655676.html,/dali
蛋白质二级结构预测
基于序列进行蛋白质二级结构方面已有了大量文献描述,本质上,这些研究可被分为两大类:基于单一序列的分析和基于多重序列对齐的分析。
文献报道PHD程序是目前此方面的最好程序,提供了从二级结构到折叠方面分析的多种资源。其网址为www.embl-heidel-berg.de/predictprotein/predictprotein.html,也可通过
email:predictprotein@embl-heidelberg.de进行数据分析。
蛋白质三级结构预测
蛋白质同源家庭的分析对于确立物种之间的亲缘关系和预测新蛋白质序列的功能有重要意义,同源蛋白质(homolog)进一步划分为直系同源(ortholog)和旁系同源(paralog),
前者指不同物种中具有相同功能和共同起源的基因,后者则指在同一物种内具有不同功能,但也有共同起源的基因,例如同是起源于珠蛋白的α珠蛋白、β珠蛋白和肌红蛋白。
蛋白质分类数据库(ProtoMap)
是对SWISS-PROT数据库中的全部蛋白质由计算机自动时行层次分类,把相关者聚集分极所得到的数据库。www.proteinmap.cs.huji.ac.il
蛋白质序列多重对齐分析及进化分析
如果发现一个未知蛋白质序列和较多不同和种属或同一种属的蛋白质序列具有较高的同源性(大于30%)那么提示待分析的蛋白质序列可能是相应家族的成员,从而可从分子时化的角度对蛋白质序列进行综合分析。
三种分析蛋白结构域的方法
三种分析蛋白结构域(Domains)的方法 1,SMART入门,蛋白结构和功能分析 SMART介绍 SMART (a Simple Modular Architecture Research Tool) allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 500 domain families found in signalling, extracellular and chromatin-associated proteins are detectable. These domains are extensively annotated with respect to phyletic distributions, functional class, tertiary structures and functionally important residues. Each domain found in a non-redundant protein database as well as search parameters and taxonomic information are stored in a relational database system. User interfaces to this database allow searches for proteins containing specific combinations of domains in defined taxa. For all the details, please refer to the publications on SMART. SMART(,可以说是蛋白结构预测和功能分析的工具集合。简单点说,就是 集合了一些工具,可以预测蛋白的一些二级结构。如跨膜区(Transmembrane segments),复合螺旋区(coiled coil regions),信号肽(Signal peptides),蛋白结构域(PFAM domains)等。 SMART前该知道的 1,SMART有两种不同的模式:normal 或genomic 主要是用的数据库不一样。Normal SMART, 用的数据库 Swiss-Prot, SP-TrEMBL 和 stable Ensembl proteomes。Genomic SMART, 用全基因组序列。详细列表:,一些名词解释 进行时 可以直接用各个数据库蛋白的ID。如Uniprot/Ensembl??ID / Accession number (ACC)。或是直接蛋白序列。运行SMART也可选择signal peptides、PFAM domains等的预测,勾上就是。看下图 SMART结果 运行后的结果用图表表示。其实运行后的结果都有明确的解释。详细请看下面。
(整理)6种方法测定蛋白质含量.
6种方法测定蛋白质含量 一、微量凯氏(kjeldahl)定氮法 样品与浓硫酸共热。含氮有机物即分解产生氨(消化),氨又与硫酸作用,变成硫酸氨。经强碱碱化使之分解放出氨,借蒸汽将氨蒸至酸液中,根据此酸液被中和的程度可计算得样品之氮含量。若以甘氨酸为例,其反应式如下: NH2CH2COOH+3H2SO4――2CO2+3SO2+4H2O+NH3(1) 2NH3+H2SO4――(NH4)2 SO4(2) (NH4)2 SO4+2NaOH――2H2O+Na2SO4+2NH3(3) 反应(1)、(2)在凯氏瓶内完成,反应(3)在凯氏蒸馏装置中进行。 为了加速消化,可以加入CuSO4作催化剂,K2SO4以提高溶液的沸点。收集氨可用硼酸溶液,滴定则用强酸。实验和计算方法这里从略。 计算所得结果为样品总氮量,如欲求得样品中蛋白含量,应将总氮量减去非蛋白 氮即得。如欲进一步求得样品中蛋白质的含量,即用样品中蛋白氮乘以6.25即得。 二、双缩脲法(biuret法) (一)实验原理 双缩脲(NH3CONHCONH3)是两个分子脲经180℃左右加热,放出一个分子氨后得到的产物。在强碱性溶液中,双缩脲与CuSO4形成紫色络合物,称为双缩脲反应。凡具有两个酰胺基或两个直接连接的肽键,或能过一个中间碳原子相连的肽键,这类化合物都有双缩脲反应。
紫色络合物颜色的深浅与蛋白质浓度成正比,而与蛋白质分子量及氨基酸成分无关,故可用来测定蛋白质含量。测定范围为1-10mg蛋白质。干扰这一测定的物质主要有:硫酸铵、tris缓冲液和某些氨基酸等。 此法的优点是较快速,不同的蛋白质产生颜色的深浅相近,以及干扰物质少。主要的缺点是灵敏度差。因此双缩脲法常用于需要快速,但并不需要十分精确的蛋白质测定。 (二)试剂与器材 1.试剂: (1)标准蛋白质溶液:用标准的结晶牛血清清蛋白(bsa)或标准酪蛋白,配制成10mg/ml的标准蛋白溶液,可用bsa浓度1mg/ml的a280为0.66来校正其纯度。如有需要,标准蛋白质还可预先用微量凯氏定氮法测定蛋白氮含量,计算出其纯度,再根据其纯度,称量配制成标准蛋白质溶液。牛血清清蛋白用H2O 或0.9%NaCl配制,酪蛋白用0.05NaOH配制。 (2)双缩脲试剂:称以1.50克硫酸铜(CuSO4?5H2O)和6.0克酒石酸钾钠(KNaC4H4O6?4H2O),用500毫升水溶解,在搅拌下加入300毫升10% NaOH溶液,用水稀释到1升,贮存于塑料瓶中(或内壁涂以石蜡的瓶中)。此试剂可长期保存。若贮存瓶中有黑色沉淀出现,则需要重新配制。 2.器材: 可见光分光光度计、大试管15支、旋涡混合器等。 (三)操作方法 1.标准曲线的测定:取12支试管分两组,分别加入0,0.2,0.4,0.6,0.8,1.0毫升的标准蛋白质溶液,用水补足到1毫升,然后加入4毫升双缩脲试剂。充分摇匀后,在室温(20~25℃)下放置30分
蛋白质定量检测方法
Bradford法蛋白定量(Bradford Protein Assay ) Bradford Assay is a rapid and accurate method commonly used to determine the total protein concentration of a sample. The assay is based on the observation that the absorbance maximum for an acidic solution of Coomassie Brilliant Blue G-250 shifts from 465 nm to 595 nm when binding to protein occurs. Both hydrophobic and ionic interactions stabilize the anionic form of the dye, causing a visible color change. Within the linear range of the assay (~5-25 mcg/mL), the more protein present, the more Coomassie binds. Reference Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. (1976) 72, 248-254. 考马斯亮蓝染色法(Bradford法)测定蛋白质含量 原理 1976年Bradford建立了用考马斯亮蓝G250与蛋白质结合的原理,迅速、敏感的定量测定蛋白质的方法。染料与蛋白质结合后引起染料最大吸收的改变,从465nm变为595nm,光吸收增加。蛋白质-染料复合物具有高的消光系数,因此大大提高了蛋白质测定的灵敏度,最低检出量为1μg蛋白。染料与蛋白质的结合是很迅速的过程,大约需2min,结合物的颜色在1h内是稳定的。一些阳离子,如K+,Na+,Mg2+,(NH4)2SO4,乙醇等物质不干扰测定,而大量的去污剂如TritonX100,SDS等严重干扰测定,少量的去污剂可通过用适当的对照而消除。由于染色法简单迅速,干扰物质少,灵敏度高,现已广泛应用于蛋白质含量的测定。 操作 一、标准方法 取含10~100μg蛋白质溶液于小试管中,用双蒸水或缓冲液调体积到0.1mL,然后加入5mL蛋白试剂,充分振荡混合,2min后于595nm测定光吸收值。以0.1mL 双蒸水或缓冲液及5mL蛋白试剂作为空白对照。 二、微量蛋白分析法 取含1~10μg蛋白质溶液,用双蒸水调体积到0.8mL,加0.2mL蛋白试剂,充分振荡混合,2min后于595nm测定光吸收值,以0.8mL双蒸水及0.2mL蛋白试剂作为空白对照。用不同浓度的蛋白质溶液作标准曲线,以蛋白质浓度为横坐
蛋白质结构解析的方法对比综述 (1)
蛋白质结构解析的方法对比综述 工程硕士李瑾 摘要:到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR法,这两种方法各有优点和不足。 关键词:x射线衍射法 NMR法 到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR法。其中X射线的方法产生的更早,也更加的成熟,解析的数量也更多,第一个解析的蛋白的结构,就是用x晶体衍射的方法解析的。而NMR方法则是在90年代才成熟并发展起来的。这两种方法各有优点和不足[1]。 首先是X射线晶体衍射法。该方法的前提是要得到蛋白质的晶体。通常是将表达目的蛋白的基因经PCR扩增后克隆到一种表达载体中,然后转入大肠杆菌中诱导表达,目的蛋白提纯之后摸索结晶条件,等拿到晶体之后,将晶体进行x射线衍射,收集衍射图谱,通过一系列的计算,得到蛋白质的原子结构[2]。 x射线晶体衍射法的优点是:速度快,通常只要拿到晶体,最快当天就能得出结构,另外不受肽链大小限制,无论是多大分子量的蛋白质或者RNA、DNA,甚至是结合多种小分子的复合体,只要能够结晶就能够得到其原子结构。所以x射线方法解析蛋白的关键是摸索蛋白结晶的条件。该方法得到的是蛋白质分子在晶体状态下的空间结构,这种结构与蛋白质分子在生物细胞内的本来结构有较大的差别。晶体中的蛋白质分子相互间是有规律地、紧密地排列在一起的,运动性较差;而自然界的生物细胞中的蛋白质分子则是处于一种溶液状态,周围是水分子和其他的生物分子,具有很好的运动性。而且,有些蛋白质只能稳定地存在于溶液状态,无法结晶[2]。 核磁共振NMR(nuclear magnetic resonance)现象很早就被科研人员观察到了,但将这种方法用来解析蛋白质结构,却是近一二十年的事情。NMR法具体原理是对水溶液中的蛋白质样品测定一系列不同的二维核磁共振图谱,然后根据已确定的蛋白质分子的一级结构,通过对各种二维核磁共振图谱的比较和解析,在图谱上找到各个序列号氨基酸上的各种氢原子所对应的峰。有了这些被指认的峰,就可以根据这些峰在核磁共振谱图上所呈现的相互之间的关系得到它们所对应的氢原子之间的距离。[3]可以想象,正是因为蛋白质分子具有空间结构,在序列上相差甚远的两个氨基酸有可能在空间距离上是很近的,它们所含的氢原子所对应的NMR峰之间就会有相关信号出现[4] 。通常,如果两个氢原子之间距离小于0.5纳米的话,它们之间就会有相关信号出现。一个由几十个氨基酸残基组成的蛋白质分子可以得到几百个甚至几千个这样与距离有关的信号,按照信号的强弱把它们转换成对应的氢原子之间的距离,然后运用计算机程序根据所得到的距离条件模拟出该蛋白质分子的空间结构。该结构既要满足从核磁共振图谱上得到的所有距离条件,还要满足化学上有关原子与原子结合的一些基本限制条件,如原子间的化学键长、键角和原子半径等[4]。 NMR解析蛋白结构常规步骤如下:首先通过基因工程的方法,得到提纯的目的蛋白,在蛋白质稳定的条件下,将未聚合,而且折叠良好的蛋白样品(通常是1mM-3mM,500ul,PH6-7的PBS)装入核磁管中,放入核磁谱仪中,然后由写好的程序控制谱仪,发出一系列的电磁波,激发蛋白中的H、13N、13C原子,等电磁波发射完毕,再收集受激发的原子所放出的“能量”,通过收集数据、谱图处理、电脑计算从而得到蛋白的原子结构[5] [6]。 用NMR研究蛋白质结构的方法,可以在溶液状态进行研究,得到的是蛋白质分子在溶液中的结构,这更接近于蛋白质在生物细胞中的自然状态[7]。此外,通过改变溶液的性质,还可以模拟出生物细胞内的各种生理条件,即蛋白质分子所处的各种环境,以观察这些周围环境的变化对蛋白质分子空间结构的影响。在溶液环境中,蛋白质分子具有与自然环境中类
测量蛋白质三种方法测定原理
测定蛋白质含量的三种方法原理 院系:xxx 专业:xxx 姓名:xxx 学号:xxx
测量蛋白质三种方法测定原理 【摘要】了解测量蛋白质三种测定方法的基本原理和优缺点。三种方法为考马斯亮蓝法(Bradford法),Folin-酚试剂法(Lowry法)和紫外吸收法。 【关键词】蛋白质含量测定考马斯亮蓝法 Folin-酚试剂法紫外吸收法 蛋白质含量测定法,是生物化学研究中最常用、最基本的分析方法之一目前常用的有两种古老的经典方法,即Folin-酚试剂法(Lowry法)和紫外吸收法。另外还有一种近十年才普遍使用起来的新的测定法,即考马斯亮蓝法(Bradford 法)。其中Bradford法和Lowry法灵敏度最高,比紫外吸收法灵敏10~20倍。。每种测定法都不是完美无缺的,都有其优缺点。在选择方法时应考虑:①实验对测定所要求的灵敏度和精确度;②蛋白质的性质;③溶液中存在的干扰物质;④测定所要花费的时间。简单列表为: 考马斯亮蓝法 双缩脲法(Biuret法)和Folin—酚试剂法(Lowry法)的明显缺点和许多限制,促使科学家们去寻找更好的蛋白质溶液测定的方法。 1976年由Bradford建立的考马斯亮兰法(Bradford法),是根据蛋白质与染料相结合的原理设计的。这种蛋白质测定法具有超过其他几种方法的突出优点,因而正在得到广泛的应用。这一方法是目前灵敏度最高的蛋白质测定法。它是一种蛋白定量法 (一)实验原理 考马斯亮蓝G-250(Coomassie G-250)是一种甲基取代的三苯基甲烷,分子中磺酸基的蓝色染料,在465nm处有最大吸收值。考马斯亮蓝G-250能与蛋白质通过范得华相互作用形成蛋白质—考马斯亮蓝复合物蓝色溶液,引起该染料的最大吸收λmax的位置发生红移,在595nm处有最大吸收值。由于蛋白质—考马斯亮蓝复合物在595nm处的光吸收远高于考马斯亮蓝在465nm处的光吸收,因此,可大大地提高蛋白质的测定灵敏度。蛋白质—考马斯亮蓝复合物溶液颜色的深浅与蛋白质的浓度成正比。利用溶液颜色的差异进行比色测定,适合于蛋白质类的定量分析,尤其适合于稀有蛋白质的微量分析。 (二)Bradford法的突出优点是: (1)灵敏度高,据估计比Lowry法约高四倍,其最低蛋白质检测量可达1mg。这
蛋白质结构分析原理及工具-文献综述
蛋白质结构分析原理及工具 (南京农业大学生命科学学院生命基地111班) 摘要:本文主要从相似性检测、一级结构、二级结构、三维结构、跨膜域等方面从原理到方法再到工具,系统地介绍了蛋白质结构分析的常用方法。文章侧重于工具的列举,并没有对原理和方法做详细的介绍。文章还列举了蛋白质分析中常用的数据库。 关键词:蛋白质;结构预测;跨膜域;保守结构域 1 蛋白质相似性检测 蛋白质数据库。由一个物种分化而来的不同序列倾向于有相似的结构和功能。物种分化后形成的同源序列称直系同源,它们通常具有相似的功能;由基因复制而来的序列称为旁系同源,它们通常有不同的功能[1]。因此,推测全新蛋白质功能的第一步是将它的序列与进化上相关的已知结构和功能的蛋白质序列比较。表一列出了常用的蛋白质序列数据库和它们的特点。 表一常用蛋白质数据库 网址可能有更新 氨基酸替代模型。进化过程中,一种氨基酸残基会有向另一种氨基酸残基变化的倾向。氨基酸替代模型可用来估计氨基酸替换的速率。目前常用的替代模型有Point Accepted Mutation (PAM)矩阵、BLOck SUbstitution Matrix (BLOSUM)矩阵[2]、JTT模型[3]。 序列相似性搜索工具。序列相似性搜索又分为成对序列相似性搜索和多序列相似性搜索。成对序列相似性搜索通过搜索序列数据库从而找到与查询序列相似的序列。分为局部联配和全局联配。常用的局部联配工具有BLAST和SSEARCH,它们使用了Smith-Waterman 算法。全局联配工具有FASTA和GGSEARCH,基于Needleman-Wunsch算法。多序列相似性搜索常用于构建系统发育树,这里不阐述。表二列举了常用的成对序列相似性比对搜索工具
几种测蛋白含量方法的比较
蛋白质含量测定方法的比较及肽含量的测定 (一)蛋白质测定方法的比较(原理、优缺点)蛋白质含量测定法,目前包括定氮法、双缩脲法、福林酚法(Lowry 法)和紫 外吸收法、考马斯亮蓝法。其中考马斯亮蓝和福林酚法灵敏度最高,比紫外吸收法灵敏10~20 倍,比双缩脲法灵敏100倍以上。定氮法较复杂,但准确,往往以定氮法测定的蛋白质作为其他方法的标准蛋白质。在选择方法时应该考虑:(1)实验测定要求的灵敏度和精确度;(2)蛋白质的性质;(3)溶液中存在的干扰物质;(4)测定花费时间。蛋白质含量测定法,目前包括定氮法、双缩脲法、福林酚法(Lowry 法)和紫外吸收法、考马斯亮蓝法。其中考马斯亮蓝和福林酚法灵敏度最高,比紫外吸收法灵敏10~20 倍,比双缩脲法灵敏100倍以上。定氮法较复杂,但准确,往往以定氮法测定的蛋白质作为其他方法的标准蛋白质。在选择方法时应该考虑:(1)实验测定要求的灵敏度和精确度;(2)蛋白质的性质;(3)溶液中存在的干扰物质;(4)测定花费时间。 1 微量凯氏定氮法(GB 5009.5-2010) 1.1原理样品与浓硫酸共热。含氮有机物即分解产生氨(消化),氨又与硫酸作用,变成硫酸氨。经强碱碱化使之分解放出氨,借蒸汽将氨蒸至酸液中,根据此酸液被中和的程度可计算得样品之氮含量。 1.2操作方法样品经前处理、炭化、消化、蒸馏、滴定等主要步骤 1.3特点准确度较高,适用于0.2~ I.Omg氮,误差为土2%;操作复杂费时,整个过程需要耗时8~10h,试剂消耗量大。,测得结果为总氮含量,包括蛋白氮和非蛋白氮含 量;适用范围广,几乎所有样品均可用此方法。 2双缩脲比色法
定量蛋白质组学的方法有哪些 (2).doc
定量蛋白质组学的方法有哪些? 1背景和意义 从生命活动的直接执行者——蛋白质的角度研究生命现象和规律(特别是疾病防治和病 理研究)已成为研究生命科学的主要手段。而这些研究往往离不开对细胞、组织或器官中含有蛋白质种类和表达量的研究。对处不同时期、不同条件下蛋白质表达水平变化的研究,识别功能模块和路径,监控疾病的生物标志物,这些研究都需要对蛋白质进行鉴定和定量。生物质谱技术的出现和不断成熟为蛋白质差异表达分析提供了更可靠、动态范围更广的研究手段。基于质谱技术,科学家们不断开发出新的定量蛋白质组学方法,来了解细胞、组织或生物体的整体蛋白质动力学。 2方法学介绍 目前较主流的定量蛋白质组学方法有 5 种,分别是Label-free 、 iTRAQ 、 SILAC 、 MRM(MRM HR) 、和 SWATH 。简述如下: 2.1Label-free Label-free 定量,即非标记的定量蛋白质组学,不需要对比较样本做特定标记处理,只 需要比较特定肽段 /蛋白在不同样品间的色谱质谱响应信号便可得到样品间蛋白表达量的变 化,通常用于分析大规模蛋白鉴定和定量时所产生的质谱数据。 Label-free 操作简单,可以做任意样本的总蛋白质差异定量,但对实验操作的稳定性、 重复性要求较高,准确性也较标记定量差。因此,Label-free 技术适合于大样本量的定量比较,以及对无法用标记定量实现的实验设计。 2.2 iTRAQ iTRAQ 定量是目前定量蛋白质组学应用很广泛的技术,该技术的核心原理是多肽标记 和定量,将多肽的含量转化为 114、115、116 和 117 同位素的含量 (或 113、114、115、116、117、118、119 和121 的 8 标记 ),从而简化了定量的复杂性,最终通过多肽定量值回归到蛋 白的定量值,从而最终测定出不同样本之间蛋白质的差异。 iTRAQ定量不依赖样本,可检测出较低丰度蛋白,胞浆蛋白、膜蛋白、核蛋白、胞外 蛋白等,且定量准确,可同时对8 个样本进行分析,并可同时得出鉴定和定量的结果,特别 适用于采用多种处理方式或来自多个处理时间的样本的差异蛋白分析。金开瑞质谱平台应用 iTRAQ 定量技术,可鉴定多达 6000 种蛋白(以人 HepG2 为例),重复样品间的蛋白表达量 相关性可达到0.95 以上。 2.3SILAC SILAC 定量的基本原理是分别用天然同位素(轻型 )或稳定同位素(中性或重型 )标记的必 需氨基酸取代细胞培养基中相应氨基酸,细胞经 5-6 个倍增周期后,稳定同位素标记的氨基 酸完全掺入到细胞新合成的蛋白质中替代了原有氨基酸。不同标记细胞的裂解蛋白按细胞数或蛋 白量等比例混合,经分离、纯化后进行质谱鉴定,根据一级质谱图中两个同位素型肽段的面积 比较进行相对定量,属于体内代谢标记法。 SILAC 属于体内标记技术,更接近样品真实状态,标记效率高达100% ,且标记效果稳 1
常见蛋白质测定方法的总结与比较
分析化学 结课作业 常见蛋白质测定方法的总结与比较 材料科学与技术学院 林化13-1班 刘旺衢 130534106
常见蛋白质测定方法的总结与比较 刘旺衢 (北京林业大学材料科学与技术学院林化13-1班 130534106,10083) 蛋白质是构成生物体细胞组织的重要成分。食物中的蛋白质是人体中氮的唯一来源。具有糖类和脂肪不可替代的作用。蛋白质与营养代谢、细胞结构、酶、激素、病毒、免疫、物质运转、遗传等密切相关,是对人类最重要的物质之一。准确精密的测定蛋白质,关乎人类的生产、生活、生存。目前测定蛋白质含量的方法有多种,如凯氏定氮法、紫外吸收法、双缩脲法、考马斯亮蓝染色法、酚试剂法等几种方法,下面本文将总结比较这五种蛋白质的测定方法。 一、凯氏定氮法 凯氏定氮法是测定化合物或混合物中总氮量的一种方法。即在有催化剂的条件下,用浓硫酸消化样品将有机氮都转变成无机铵盐,然后在碱性条件下将铵盐转化为氨,随水蒸气馏出并为过量的酸液吸收,再以标准酸滴定,就可计算出样品中的氮量。由于蛋白质含氮量比较恒定,可由其氮量计算蛋白质含量,故此法是经典的蛋白质定量方法。蛋白质是含氮的有机化合物。蛋白质与硫酸和催化剂一同加热消化,使蛋白质分解,分解的氨与硫酸结合生成硫酸铵。然后碱化蒸馏使氨游离,用硼酸吸收后再以硫酸或盐酸标准溶液滴定,根据酸的消耗量乘以换算系数计算蛋白质含量,即含氮量*6.25=蛋白含量。 凯氏定氮法具有灵敏度高, 样品用量少,最低可检出0.05mg氮;精密度、准确度高,平行误差一般小于0.5%;应用范围广,适用于一切形态的食品与生物样品;仪器装置简单,试剂廉价的优点。 但也存在操作比较繁琐费时,特别是蒸馏定氮过程的效率低,不利于大批样品的测定;定氮的结果既包括有机氮,也包括无机氮,有机氮中除蛋白氮外,还包括非蛋白氮,测定的结果只能是粗蛋白质的含量;在蛋白质氨基酸构成有差异的
三种定量蛋白质组方法比较
A comparative study on the three quantitative methods frequently used in proteomics, 2D DIGE (difference gel electrophoresis), cICAT (cleavable isotope-coded affinity tags) and iTRAQ (isobaric tags for relative and absolute quantification), was carried out. DIGE and cICAT are familiar techniques used in gel- and LC-based quantitative proteomics, respectively. iTRAQ is a new LC-based technique which is gradually gaining in popularity. A systematic comparison among these quantitative methods has not been reported. In this study, we conducted well-designed comparisons using a six-protein mixture, a reconstituted protein mixture (BSA spiked into human plasma devoid of(缺乏)six abundant proteins), and complex HCT-116 cell lysates as the samples. All three techniques yielded quantitative results with reasonable accuracy when the six-protein or the reconstituted protein mixture was used. In DIGE, accurate quantification was sometimes compromised due to comigration or partial comigration of proteins. The iTRAQ method is more susceptible to errors in precursor ion isolation, which could be manifested with increasing sample complexity. The quantification sensitivity of each method was estimated by the number of peptides detected for each protein. In this regard, the global-tagging iTRAQ technique was more sensitive than the cysteine-specific cICAT method, which in turn was as sensitive as, if not more sensitive than, the DIGE technique. Protein profiling on HCT-116 and HCT-116 p53 ?/? cell lysates displayed limited overlapping among proteins identified by the three methods, suggesting the complementary nature of these methods. Keywords: protein quantification ? DIGE ? cICAT ? iTRAQ ? 2D gel ? LC-MALDI TOF/TOF DIGE:荧光差异凝胶电泳 CICAT:同位素亲和标记 iTRAQ:同位素标记相对和绝对定量
蛋白质各种定量方法的优缺点的比较
1.蛋白质的常规检测方法 凯氏(Kjeldahl )定氮法 一种最经典的蛋白质检测方法。 原理:样品中含氮有机化合物与浓硫酸在催化剂作用下共热消化, 含氮有机物分解产生氨, 氨又与硫酸作用变成硫酸铵。然后加碱蒸馏放出氨, 氨用过量的硼酸溶液吸收, 再用盐酸标准溶液滴定求出总氮量换算为蛋白质含量。 优点:范围广泛、测定结果准确、重现性好 缺点:操作复杂费时、试剂消耗量大 双缩脲法 常用于需要快速但并不需要十分精确的蛋白质检测。 原理:双缩脲(NHCONHCONH是3分子的脲经180C左右加热,放出1分子氨后得到的产物。在强碱性溶液中,双缩脲与硫酸铜形成紫色络合物(肽键中的氮原子和铜离子配价结合),称为双缩脲反应。紫色络合物颜色的深浅与蛋白质浓度成正比,因此可用来测定蛋白质含量。 测定范围:1~10mg(有的文献记载为1~20mg) 优点:较快速,干扰物质少,不同蛋白质产生的颜色深浅相近 缺点:①灵敏度差; ② 三羟甲基氨基甲烷、一些氨基酸和EDTA等会干扰该反应。 Folin- 酚试剂法 原理:Folin- 酚法的原理与双缩脲法大体相同,利用蛋白质中的肽键与铜结合产生双缩脲反应。同时也由于Folin- 酚试剂中的磷钼酸- 磷钨酸试剂被蛋白质中的酪氨酸和苯丙氨酸残基还原,产生深蓝色的钼蓝和钨蓝的混合物。在一定的条件下, 蓝色深度与蛋白的量成正比,由此可测定蛋白质的含量。
测定范围:20~250ug 优点:灵敏度高,对水溶性蛋白质含量的测定很有效 缺点:①费时,要精确控制操作时间; ②Folin -酚法试剂的配制比较繁琐,且酚类和柠檬酸、硫酸铵、Tris缓冲液、甘氨 酸、糖类、甘油、还原剂(二硫代苏糖醇、巯基乙醇)、EDTA和脲素均会干扰反应。 紫外吸收法 原理:蛋白质分子中的酪氨酸、苯丙氨酸和色氨酸残基使其在280nm 处具有紫外吸收,其吸光度与蛋白质含量成正比)。此外,蛋白质溶液在280nm的吸光度值与肽键含量成正比,利用一定波长下蛋白质溶液的吸光度值与蛋白质浓度的正比关系可以测定蛋白质含量。 优点:简便、灵敏、快速,不消耗样品,测定后能回收。 缺点:①测定蛋白质含量的准确度较差,专一性差; ②干扰物质多,若样品中含有嘌呤、嘧啶及核酸等能吸收紫外光的物质,会出现较 大的干扰。 定氮法、双缩脲法、Filon- 酚试剂法和紫外吸收法为常用的 4 种古老的经典方法。 1.5 考马斯亮蓝法 原理:染料考马斯亮蓝G-250 在酸性溶液中与蛋白质中的碱性氨基酸(特别是精氨酸)及芳香族氨基酸残基相结合,使染料最大吸收峰的位置由465nm变为595nm,溶液的颜色也由棕黑色变为蓝色,在595nm下测定的吸光度值与蛋白质浓度呈正比。 优点:灵敏度高,测定快速、简便,干扰物质少,不受酚类、游离氨基酸和缓冲剂、络合剂的影响,适合大量样品的测定。 缺点:由于各种蛋白质中的精氨酸和芳香族氨基酸的含量不同有较大的偏 ,因此用于不同蛋白质测定时 差。
蛋白质定量的五种方法
蛋白质定量的五种方法 令狐采学 方法一双缩脲法测定蛋白质浓度 [目的]掌握双缩脲法测定蛋白质浓度的原理和标准曲线的绘制。 [原理] 双缩脲(NH2CONHCONH2)在碱性溶液中与硫酸铜反应生成紫红色化合物,称为双缩脲反应,蛋白质分子中含有许多肽键(-CONH-)在碱性溶液中也能与Cu2+反应产生紫红色化合物。在一定范围内,其颜色的深浅与蛋白质浓度成正比。因此,可以利用比色法测定蛋白质浓度。 双缩脲法是测定蛋白质浓度的常用方法之一。操作简便、迅速、受蛋白质种类性质的影响较小,但灵敏度较差,而且特异性不高。除-CONH-有此反应外,-CONH2、-CH2NH2、-CS-NH2等基团也有此反应。 [操作] 取中试管7支,按下表操作。
各管混匀、放置37℃水浴中保温20分钟。用540nm比色,以空白管调零点,读取各管光密度值。 [计算] (一)在座标纸上以光密度为纵座标,以蛋白质浓度为横座标绘制标准曲线。 (二)从标准曲线中查出待测血清样本的蛋白质浓度(g/L),并求出人血清样本的蛋白质 浓度。 (三)再从标准管中选择一管与测定管光密度相接近者,求出人血清样本的蛋白质浓度(g/L)。 [器材] 中试管7支,l毫升刻度吸管3支,10毫升刻度吸管1支,水浴箱,721型分光光度计、坐标纸。 [试剂] (—)6N NaOH:称取240g氢氧化钠溶于1000ml水中。 (二)双缩脲试剂:称取CuS04·5H2O 3.0克,酒石酸钾9.0 克和碘化钾5.0克,分别溶解后混匀,加6N NaOH l00ml,最后加
水至1000ml,贮于棕色瓶中,避光,可长期保存。如有暗红色沉淀出现,即不能使用。 (三)0.9%NaCl。 (四)蛋白质标准液(10mg/m1),称取干燥的牛血清蛋白100.0mg,以少量生理盐水溶解后倒入l0ml容量瓶中,淋洗称量瓶数次,一并倒入容量瓶中,最后加生理盐水至刻度线,或用凯氏定氮法测定血清蛋白质含量,然后稀释成l0mg/m1作为蛋白质标准液。 (五)待测血清样本:将人血清或动物血清用生理盐水稀释10倍后再测定。 方案二Folin-酚试剂法(Lowry法)测定蛋白质浓度 [目的]掌握Lowry法测定蛋白质浓度的原理。 [原理] 蛋白质在碱性溶液中其肽键与Cu2+螯合,形成蛋白质一铜复合物,此复合物使酚试剂的磷钼酸还原,产生蓝色化合物,在一定条件下,利用蓝色深浅与蛋白质浓度的线性关系作标准曲线并测定样品中蛋白质的浓度。 [操作] 取试管7支、编号、按下表操作:
蛋白质检测方法汇总
蛋白质检测方法汇总 2010-04-26 12:00:38| 分类:蛋白电泳| 标签:|字号大中小订阅 本文引用自啸月天狼《蛋白质检测方法汇总》 更多相关资料请查看https://www.360docs.net/doc/929655676.html, 蛋白质检测方法汇总 本实验的目的是学会各种蛋白质含量的测定方法。 了解各种测定方法的基本原理和优缺点。 蛋白质含量测定法,是生物化学研究中最常用、最基本的分析方法之一。目前常用的有四种古老的经典方法,即定氮法,双缩尿法(Biuret法)、Folin-酚试剂法(Lowry法)和紫外吸收法。另外还有一种近十年才普遍使用起来的新的测定法,即考马斯亮蓝法(Bradford 法)。其中Bradford法和Lowry法灵敏度最高,比紫外吸收法灵敏10~20倍,比Biuret法灵敏100倍以上。定氮法虽然比较复杂,但较准确,往往以定氮法测定的蛋白质作为其他方法的标准蛋白质。 值得注意的是,这后四种方法并不能在任何条件下适用于任何形式的蛋白质,因为一种蛋白质溶液用这四种方法测定,有可能得出四种不同的结果。每种测定法都不是完美无缺的,都有其优缺点。 在选择方法时应考虑: ①实验对测定所要求的灵敏度和精确度; ②蛋白质的性质; ③溶液中存在的干扰物质; ④测定所要花费的时间。 考马斯亮蓝法(Bradford法),由于其突出的优点,正得到越来越广泛的应用。 一、微量凯氏(Kjeldahl)定氮法 样品与浓硫酸共热。含氮有机物即分解产生氨(消化),氨又与硫酸作用,变成硫酸氨。经强碱碱化使之分解放出氨,借蒸汽将氨蒸至酸液中,根据此酸液被中和的程度可计算得样品之氮含量。若以甘氨酸为例,其反应式如下: CH2COOH | + 3H2SO4--------- 2CO2 + 3SO2 +4H2O +NH3 (1) NH2 2NH3 + H2SO4 -------- ---(NH4)2SO4 (2) (NH4)2SO4 + 2NaOH ----------- 2H2O +Na2SO4 + 2NH3 (3) 反应(1)、(2)在凯氏瓶内完成,反应(3)在凯氏蒸馏装置中进行。 为了加速消化,可以加入CuSO4作催化剂,K2SO4以提高溶液的沸点。收集氨可用硼酸溶液,滴定则用强酸。实验和计算方法这里从略。计算所得结果为样品总氮量,如欲求得样品中蛋白含量,应将总氮量减去非蛋白氮即得。如欲进一步求得样品中蛋白质的含量,即用样品中蛋白氮乘以6.25即得。 五种蛋白质测定方法比较如下: 方法灵敏度时间原理干扰物质说明 凯氏定氮法(Kjedahl法)灵敏度低,适用于0.2~ 1.0mg氮,误差为±2%费时8~10小时将蛋白氮转化为氨,用酸吸收后滴定非蛋白氮(可用三氯乙酸沉淀蛋白质而分离)用于标准蛋白质含量的准确测定;干扰少;费时太长 双缩脲法(Biuret法)灵敏度低1~20mg 中速20~30分钟多肽键+碱性Cu2+?紫色络合物硫酸铵;Tris缓冲液;某些氨基酸用于快速测定,但不太灵敏;不
尿蛋白定量与定性是怎样的
尿蛋白定量与定性是怎样的 尿蛋白定量与定性是怎样的?正常人尿中含有少量蛋白质,主要为白蛋白,24小时尿中含量约为40~80毫克,最多不超过150毫克。由于量少,常规定性检查为阴性,故临床上习惯于说正常人尿液无蛋白质。当尿中蛋白质浓度超过150毫克,日时,尿常规定性检查己能将此蛋白检出,此尿即称为蛋白尿。蛋白尿是肾炎的主要表现之一。 尿蛋白定性能判断病人是否有蛋白尿,并能通过半定量分析粗略估计尿中蛋白质含量。常用的检测方法有加热醋酸法及磺基水杨酸法等。试验结果尿液若无混浊及沉淀则为阴性,以(-)表示;若出现不同程度的混浊或沉淀则为阳性,用(+)~(++++)作半定量估计。尿蛋白定性(半定量)结果与其相应尿蛋白浓度间大致对应关系已显示见表1。 1.肾小球性蛋白尿:肾小球性蛋白尿(glomerular proteinuria)是由于肾小球滤过膜对血浆蛋白通透性增高所致。是临床最多见的类型。见于多种原发或继发性肾小球肾炎。是由于缺血、中毒、免疫病理损伤破坏了滤过膜的完整性;或由于滤过膜电荷屏障作用减弱而致。 2.肾小管性蛋白尿:在正常情况下经肾小球滤出的中小分子蛋白质,几乎全被肾小管重吸收。当肾小 管疾病时,蛋白质重吸收受障碍,小分子蛋白质就会从尿中排出,包括β2-微球蛋白、溶菌酶、核糖核酸酶等。由于血液中小分子蛋白质浓度很低,故此类病人尿蛋白总量一般不超过2g,有时仅10mg,也可以蛋白尿存在。 3.溢出性蛋白尿:在某些疾病中,血中小分子蛋白浓度增加,如本周氏蛋白、血红蛋白、肌红蛋白等:若滤液中浓度超过肾吸收阈限,就可以排出,见于骨髓瘤、血管内溶血性疾病。 4.分泌性蛋白尿:尿中IgA排泄增多,见于肾小管间质性肾病;髓袢升支厚段受炎症及药物刺激时,分泌粘液蛋白(Tamm Horsfell蛋白,T-H蛋白)增多。 5.组织性蛋白尿:组织遭受破坏后可以释放胞质中各种酶及蛋白质,若分子量小,肾小球滤液中浓度超过肾小管吸收阈限,就则可自尿中排出。该项蛋白液可以直接从肾小管排出,如癌胚抗原——aFP、溶菌酶、肾小管基膜抗原等。 因此,测定蛋白质的大小及性质对临床很有意义。SDS据丙酰胺凝胶电泳的应用更有实际价值。 总之,如果你没有学过医,那我告诉你,蛋白尿只是一个症状而不是病,治疗蛋白尿关键是认识他,为什么会有蛋白尿。糖尿病高血压是不是控制好了。 尿蛋白测定,包括尿蛋白定性和尿蛋白定量。 尿蛋白定性的临床意义: 尿蛋白定性有生理性的,也有病理性,其阳性程度与肾损害程度不一定成正比。糖尿病患者尿中持续出现尿蛋白阳性,除外泌尿系感染、原发性肾病外,应考虑糖尿病肾病的诊断。有专家认为,糖尿病肾病早期蛋白尿呈间歇性,只在劳动或运动后为阳性反应。因此,运动后尿蛋白检验对诊断糖尿病肾病的早期发现有一定的意义。
5种蛋白质分析方法
蛋白质分析方法 1、微量凯氏(Kjeldahl)定氮法 样品与浓硫酸共热。含氮有机物即分解产生氨(消化),氨又与硫酸作用,变成硫酸氨。经强碱碱化使之分解放出氨,借蒸汽将氨蒸至酸液中,根据此酸液被中和的程度可计算得样品之氮含量。若以甘氨酸为例,其反应式如下: NH2CH2COOH+3H2SO4——2CO2+3SO2+4H2O+NH3 (1) 2NH3+H2SO4——(NH4)2SO4 (2) (NH4)2SO4+2NaOH——2H2O+Na2SO4+2NH3 (3) 反应(1)、(2)在凯氏瓶内完成,反应(3)在凯氏蒸馏装置中进行。 为了加速消化,可以加入CuSO4作催化剂,K2SO4以提高溶液的沸点。收集氨可用硼酸溶液,滴定则用强酸。实验和计算方法这里从略。 计算所得结果为样品总氮量,如欲求得样品中蛋白含量,应将总氮量减去非蛋白 氮即得。如欲进一步求得样品中蛋白质的含量,即用样品中蛋白氮乘以6.25即得。 评价: 总氮-非蛋白氮=蛋白质氮——>蛋白质含量 灵敏度低,误差大,耗时长。 2、双缩脲法(Biuret法) (一)实验原理 双缩脲(NH3CONHCONH3)是两个分子脲经180℃左右加热,放出一个分子氨后得到的产物。在强碱性溶液中,双缩脲与CuSO4形成紫色络合物,称为双缩脲反应。凡具有两个酰胺基或两个直接连接的肽键,或能过一个中间碳原子相连的肽键,这类化合物都有双缩脲反应。 紫色络合物颜色的深浅与蛋白质浓度成正比,而与蛋白质分子量及氨基酸成分无关,故可用来测定蛋白质含量。测定范围为1-10mg蛋白质。干扰这一测定的物质主要有:硫酸铵、Tris 缓冲液和某些氨基酸等。 此法的优点是较快速,不同的蛋白质产生颜色的深浅相近,以及干扰物质少。主要的缺点是灵敏度差。因此双缩脲法常用于需要快速,但并不需要十分精确的蛋白质测定。(二)试剂与器材 1. 试剂: (1)标准蛋白质溶液:用标准的结晶牛血清清蛋白(BSA)或标准酪蛋白,配制成10mg/ml 的标准蛋白溶液,可用BSA浓度1mg/ml的A280为0.66来校正其纯度。如有需要,标准蛋白质还可预先用微量凯氏定氮法测定蛋白氮含量,计算出其纯度,再根据其纯度,称量配制成标准蛋白质溶液。牛血清清蛋白用H2O 或0.9%NaCl配制,酪蛋白用0.05N NaOH 配制。 (2)双缩脲试剂:称以 1.50克硫酸铜(CuSO4?5H2O)和 6.0克酒石酸钾钠(KNaC4H4O6?4H2O),用500毫升水溶解,在搅拌下加入300毫升10% NaOH溶液,用水稀释到1升,贮存于塑料瓶中(或内壁涂以石蜡的瓶中)。此试剂可长期保存。若贮存瓶中有黑色沉淀出现,则需要重新配制。
尿液蛋白质定量测定方法及临床意义
尿液蛋白质定量测定方法及临床意义 【摘要】正常人尿液中含有微量的蛋白质,每天排出量在150mg 以下,其中以白蛋白占多数,随不同病种有一定量的球蛋白和其它蛋白。随着肾病科研工作的开展,以观察患者每天排出蛋白质的量以示病情的变化,因此尿液蛋白质定量在临床上有实际应用价值,已成为医院化验室常规的检验项目。 【关键词】尿液蛋白质检测 1丽春红-S法 尿液标本中的蛋白质与丽春红-S染料混匀后一起被三氯醋酸沉淀,将沉淀物溶解于碱性溶液中,此时溶液呈紫色,显色深度与蛋白质浓度成正比。 1.1试剂 三氯乙酸-丽春红-S试剂原液:称取丽春红-S1.0g,加入到1000ml300g/L三氯乙酸中溶解。 三氯乙酸-丽春红-S试剂应用液:将原液用蒸馏水按1:10稀释,放置室温稳定数月。 蛋白定性试剂。0.2mol/L氢氧化钠溶液。 蛋白标准曲线:把定值蛋白稀释成不同的浓度,按操作测定其吸光度,以吸光度为横坐标,以稀释后的蛋白浓度为纵坐标绘制标准曲线。 1.2操作先做尿蛋白定性试验,以适当稀释标本。在试管中加入100μl标本,再加入1ml三氯乙酸一丽春红-S应用液,混匀后以3000r
/min离心15分钟,吸去上清液,倒置于洁净滤纸上。在沉淀中加入0.2mol/L氢氧化钠溶液lml,用560nm波长测定其吸光度,查标准曲线的蛋白含量。 1.3参考值46.5±18.1mg/L 1.4注意事项:尿液如被稀释应乘以稀释倍数。尿中血红蛋白含量>5mg/L时影响结果。离心后上清必须全部弃去,若沉淀中夹带有丽春红-S影响比色结果。 2双缩脲比色法 以磷钨酸乙醇溶液沉淀尿中的蛋白质,在碱性介质中蛋白质与铜离子形成紫红色络合物,与标准液比色即可得出尿中蛋白质的含量。 2.1试剂 蛋白沉淀液:称取磷钨酸7.5g,加入30ml蒸馏水、95%乙醇385ml、浓盐酸25ml。 双缩脲液:(1)称取枸橼酸钠34.6g,无水碳酸钠20g,加入120ml 蒸馏水使其溶解;(2)称取硫酸铜3.46g,加入20ml蒸馏水使其溶解。将(1)、(2)混合,加入蒸馏水至200ml,备用。0.75mol/L氢氧化钠溶液。蛋白标准液(1.5g/L):称取150mg结晶型冻干人血清白蛋白,加入100ml的3g/L苯甲酸溶液溶解。 2.2操作取5ml24小时混合尿液,2500~3000r/min离心5分钟,取上清备检。混匀放入56℃水浴15分钟,取出后离心,倾上清留沉淀。0.75mol/1 NaOH1.51.51.5