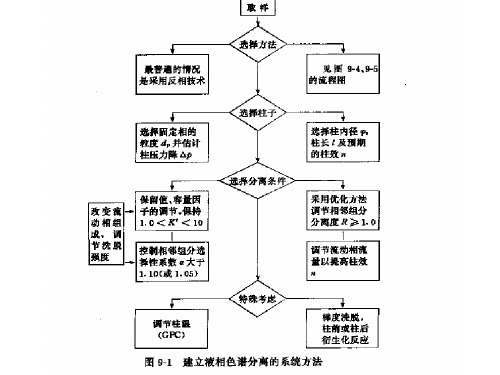
建立色谱分析方法需考查的内容
建立液相色谱方法一般步骤
液相色谱仪的使用
必须做!
溶剂和水的前处理:过滤
• 设备和材料:溶剂过滤器、真空泵、0.45mm或更 细的水性滤膜和有机滤膜
• 过滤的方法:用加了滤膜的溶剂过滤器和真空泵 抽滤
• 过滤的目的:除去溶剂中的微小颗粒,避免堵塞 色谱柱,尤其是使用无机盐配制 的缓冲液。
方法
反相 HPLC
离子对 HPLC
正相 HPLC
样品特点/色谱柱
何时应用该方法
流动相:水/有机溶剂
色谱柱:C18(ODS),C8,苯基, 三甲基硅烷,氰基
首选为能溶于水/有机混合物的中性或非 离子化合物。
流动相:水/有机溶剂(一种缓冲 物控制pH和离子对试剂)
色谱柱:C18(ODS),C8,氰基
离子或可电离化合物,尤其是碱性或阳 离子化合物的良好选择
液相色谱分析
液相色谱的检测模式
灵敏度、选择性、适应能力
项目 示差折光 电导
响应 灵敏度 线性范围
通用 毫克 104
选择性 纳克 106
流速敏感 温度敏感 梯度淋洗
是 是 不可
是 是 有限制
紫外 选择性
纳克 105 否 否 可以
荧光 选择性
皮克 105 否 否 可以
电化学 选择性
皮克 106 是 是 脉冲可以
改变容量因子 K'
①
①
②
50/50
② ③
③ 60/40
① 40/60
② ③
液相色谱分析
分析方法建立的实例
改变选择性∶a
• 通过改变流动相改变选择性
– 同样强度的不同溶剂
中国药科大学药物代谢动力学实验考查知识点整理

中国药科大学药物代谢动力学实验考查知识点整理中国药科大学药物代谢动力学实验考查知识点整理药物代谢动力学实验考查知识点整理第一部分:HPLC使用注意事项1、HPLC组成:泵、进样器、色谱柱、检测器、数据系统/积分仪2、反相色谱:分离机理:“反相色谱”固定相极性小于流动相极性常用流动相:乙腈、甲醇,水 3、色谱柱的分类:按填料:球形、无定形按含碳量:C18、C8按应用:分析柱、制备柱、预处理柱按粒径:150mm*,5μm等按填料类型:正相柱、反相柱、手性柱 4、键合相色谱柱的优缺点:优点:稳定不易流失;应用广泛,可使用多种溶剂;消除硅羟基的不良影响;缺点:pH得在3~8范围内5、C18柱的活化:90% 10% 90%的甲醇溶液1ml/min依次冲洗30min6、流动相:使用之前需超声脱气目的:色谱泵输液准确提高检测性能保护色谱柱流动相脱气的方法:加热,抽真空,超声,通惰性气体流动相组成:流动相配置:缓冲溶液现用现配,不要储存时间过长,避免pH值发生变化和成分分解,影响色谱分离的效果;有机溶液和缓冲液使用前均需经μm微孔滤膜过滤;流动相使用前脱气。
7、常用定量方法:外标法内标法内标物的要求:化学结构与待测品相似;样品中不存在;不与样品组分发生化学反应;保留值与待测值接近;浓度相当;与其他色谱峰分离好8、样品的预处理:目的:除杂质;浓缩微量成分;改善分离;保护色谱柱;提高检测灵敏度方法:高速离心,过滤,选择性沉淀,衍生反应;液固萃取、液液萃取沉淀蛋白的溶剂:有机溶剂:乙腈、甲醇强酸:三氯乙酸、过氯酸盐:50%硫酸铵、10%TCA 分析测定用试剂为色谱纯及以上,水为超纯水第二部分:实验设计1、考虑问题:动物种属、给药剂量、取血时间、采样量、血药浓度大概范围2、分析方法建立:色谱柱、流动相组成、柱温箱温度、样品处理方法、风量下限与标曲范围第三部分:实验相关条件与数据处理实验一:1、采血点设计一般原则为获得给药后的一个完整的血药浓度-时间曲线,采样时间点的设计应兼顾药物的吸收相、平衡相和消除相。
液相色谱分析方法建立-系列一基本理论及色谱介绍

硅 胶 的 溶 解 曲 线
1 2 3 4 5 6 7 8 9 p H
硅胶表面键合相的水解(PH<2)
O
OH
Si
O
+
O
“Column Bleed”
H3C
C
C
C
C
Si
C
C
C
CH3
Cl
CH3
O
+ H3C
C
C
C
C
Si
C
C
C
CH
O 硅胶表面未被键合的酸性硅羟基将会与被分析物相互作用,特别是在 中性条件下会对强碱性化合物产生严重的峰形拖尾。
O 封尾工艺使得表面残余的硅羟基被进一步的消除,为小分子所取代。 O 封尾工艺能促使色谱柱对极性和碱性化合物的分离具有良好的峰形。
50
色谱柱的pH值
O 填料的pH稳定性 O 硅胶填料 pH:2-8 O 聚合物填料 pH:2-12 O 杂化硅胶填料 pH:2-12
O
Low pH (hydrolysis of ligand)
O Si
O
O
OH
+
H3C
C
C
C
C
Si
C
C
C
CH3
HO
CH3
硅胶的溶解(PH>8)
Nucleophilic attack
• Complete dissolution of Silica
• Catastrophic column failure
蒸发光散射
任何挥发性低于流动相的样 品均能被 检测
可检测所有物质。
第四章 色谱分析法

(2)外标法
外标法也称为标准曲线法。 特点及要求: • • • 外标法不使用校正因子,准确性较高, 操作条件变化对结果准确性影响较大。 对进样量的准确性控制要求较高,适用于大批量试样的
快速分析。
(3)内标法
内标物要满足以下要求:
(a)试样中不含有该物质;
(b)与被测组分性质比较接近; (c)不与试样发生化学反应; (d)出峰位置应位于被测组分附近。
从色谱流出曲线上,可以得到许多重要信息: (1) 根据色谱峰的个数,可以判断样品中所含组 分的最少个数. (2)根据色谱峰的保留值(或位置),可以进行 定性分析. (3) 根据色谱峰的面积或峰高,可以进行定量分 析. (4)色谱峰的保留值及其区域宽度,是评价色 谱柱分离效能的依据. (5)色谱峰两峰间的距离,是评价固定相(和流 动相)选择是否合适的依据.
相同的色谱分析条件下,某组份2的调整保留值与组份 1的调整保留值之比,称为相对保留值:
2.1
VR2 tR2 t R1 VR1
由于相对保留值只与柱温及固定相的性质有关,而与柱 径、柱长、填充情况及流动相流速无关,因此,它是色 谱法中,特别是气相色谱法中,广泛使用的定性数 据. 必须注意,相对保留值绝对不是两个组份保留时 间或保留体积之比 .
⑵保留时间tR 试样从进样开始到柱后出现峰极大值时所 经历的时间,称为保留时间,如图中 O′B.它相应于样品 到达柱末端的检测器所需的时间. ⑶调整保留时间tR′某组份的保留时间扣除死时间后称为该 组份的调整保留时间,即 tR′ = tR-tM
⑷死体积 VM 指色谱柱在填充后,柱管内固定相颗粒 间所剩留的空间、色谱仪中管路和连接头间的空间 以及检测器的空间的总和.当后两项很小而可忽略 不计时,死体积可由死时间与流动相体积流速F0 (L/min)计算:
(完整word版)色谱分析方法的建立

色谱分析方法的建立第一部分色谱法系统的选择与应用第二部分气相色谱法的建立第三部分液相色谱法的建立第一部分色谱法系统的选择与应用第一章气相色谱法的特点第二章高效液相色谱法的特点第三章气相色谱和液相色谱的比较第四章分析条件的确定第五章样品的制备第六章定量第七章仪器选购的原则第二部分气相色谱法的建立第一章前言第二章色谱分离条件选择的指标第三章初始操作条件的确定第四章填充柱的选择要点第五章毛细管柱的选择第六章检测器操作条件的选择第七章程序升温操作条件的选择第三部分液相色谱法的建立第一章结论第二章分离的基础第三章检测灵敏度与选择性第四章色谱柱第五章反相HPLC第六章梯度洗脱第七章常规样品反相分离的系统方法第一部分色谱法系统的选择与应用第一章气相色谱法的特点1,色谱柱内现象单纯,可以从理论上理解.2,若用非极性柱,组分按沸点顺序流出.因此,在分配气相色谱法中,若已知化合物,则可预测流出顺序。
3,能较容易地制备具有高分离能力的色谱柱,并可在较长时间内保持稳定的性能。
4,因为移动相是气体,其粘度小,所以增加色谱柱长度可改善分离能力.5,样品组分在固定相和移动相中易扩散,能迅速达到分配平衡,故可提高移动相的流速以缩短分析时间。
6,检测惰性气体中的样品组分时,可以使用各种高灵敏检测器,所以能做极微量分析和特定组分的高灵敏度的选择性检测。
7,使用通用型检测器时,可以预测注入样品在多大程度上能作为色谱峰流出并被检测,所以分析的可信度高。
在不要求精度时,也可以把峰面积百分数近似作为组成百分数,进行快速定量分析。
8,容易与质谱仪或付里叶变换红外光谱仪联用,便于多组分混合物的分离和鉴定.9,适于分析的物质仅限于在某种形式下为热稳定的挥发性物质。
所以从全部化合物来看,适用范围较窄,约为20%。
10,用选择性检测器做微量分析时,样品必须经过前处理以除去干扰组分.第二章高效液相色谱法的特点1,适用样品范围广,80%的分析对象可借此法分析。
色谱分析方法基本理论

色谱分析方法基本理论一、保留时光理论保留时光是样品从进入色谱柱到流精彩谱柱所需要的时光,不同的物质在不同的色谱柱上以不同的流淌相洗脱会有不同的保留时光,因此保留时光是色谱分析法比较重要的参数之一。
保留时光由物质在色谱中的分配系数打算: tR=t0(1+KVs/Vm)式中:tR —某物质的保留时光; t0—色谱系统的死时光,即流淌相进入色谱柱到流精彩谱柱的时光,这个时光由色谱柱的孔隙、流淌相的流速等因素打算; K-分配系数; Vs,Vm—固定相和流淌相的体积。
这个公式又叫做色谱过程方程,是色谱学最基本的公式之一。
在薄层色谱中没有样品进入和流出固定相的过程,因此人们用比移值标示物质的色谱行为。
比移值是一个与保留时光相对应的概念,它是样品点在色谱过程中移动的距离与流淌相前沿移动距离的比值。
与保留时光一样,比移值也由物质在色谱中的分配系数打算: Rf=Vm/(Vm+KVs) 式中:Rf—比移值;K一色谱分配系数; Vs,Vm—固定相和流淌相的体积。
二、塔板理论塔板理论是色谱学的基础理论。
塔板理论将色谱柱看作一个分馏塔,待分别组分在分馏塔的塔板间移动,在每一个塔板内组分分子在固定相和流淌相之间形成平衡,随着流淌相的流淌,组分分子不断从一个塔板移动到下一个塔板并不断形成新的平衡。
色谱柱的塔板数越多,其分别效果越好。
按照塔板理论,待分别组分流精彩谱柱时的浓度随时光展现二项式分布,当色谱柱的塔板数很高时,二项式分布趋于正态分布。
流出曲线上组分浓度与时光的关系可以表示如下:式中:Ct—t时刻的组分浓度; C0—组分总浓度,即峰面积;σ—半峰宽,即正态分布的标准差; tR—组分的保留时光。
该方程称作流出曲线方程。
按照流出曲线方程,色谱柱的理论塔板高度被定义为单位柱长度的色谱峰方差: H=σ2/T 理论塔板高度越低,在单位长度色谱柱中的塔板数越多,分别效果越好。
打算理论塔板高度的因素有固定相的材质、色谱柱的匀称程度、流淌相的理化性质以及流淌相的流速等。
分光光度法、色谱法(三)

分光光度法、色谱法(三)一、最佳选择题1. 采用紫外分光光度法测定氯氮卓片的含量时,在308nm处测得供试品溶液的吸收度为0.638,已知氯氮革在308n(江南博哥)m处的吸收系数为319,则供试品溶液的浓度为A.0.005g/100mlB.0.02g/100mlC.0.005g/mlD.0.002g/mlE.0.002g/100ml`正确答案:E[解析] 本题考查吸收度的相关内容。
根据A=ECL,可知与溶液浓度和液层厚度成正比的是吸光度。
本题中浓度C=0.638/319=0.002。
故答案为E。
2. 采用紫外-可见光光度法测定对乙酰氨基酚的含量时,若稀释体积为D(ml),取样量为W(g),含量计算公式是A.含量(%)=[(A×D)/(×100×W)]×100%B.含量(%)=[(×100×W)/(A×D)]×100%C.含量(%)=[(×A×D)/(100×W)]×100%D.含量(%)=[(A×W)/(×100×D)]×100%E.含量(%)=[(×D)/(A×100×W)]×100%正确答案:A[解析] 本题考查对乙酰氨基酚的含量计算。
用紫外-可见光光度法测定,采用吸收系数法:含量%=(Ax·D)/(100·W·L·)。
故答案为A。
3. 下列溶液中,用于紫外-可见分光光度计吸光度准确度检定的是A.氢氧化钠溶液B.磷酸盐缓冲液C.盐酸溶液D.氯化钠溶液E.重铬酸钾的硫酸溶液正确答案:E[解析] 本题考查紫外-可见分光光度计吸光度准确度的检定。
我国在1992年制订的《紫外-可见分光光度计国家标准》中规定,用重铬酸钾溶液来测试紫外区的吸光度准确度,并明确指出了测试波长。
液相色谱分析方法的建立【最新】

一. 方法建立的步骤二.开始前应知道1. 样品的性质在开始方法建立之前,我们应该检查自己对样品的了解程度,并明确分离目标。
表 1 有关样品组分和性质的重要信息所含化合物的数目化合物的化学结构(官能团)化合物的分子量化合物的pKa值化合物的UV光谱图化合物在样品中的浓度范围样品的溶解度样品的化学成分能够为选择HPLC分离的最佳初始条件提供有价值的线索根据已知的样品信息,HPLC方法建立有两种不甚相同的模式。
一种模式依据样品的“化学性质”选择最佳初始条件,色谱工作者需很大程度依赖于过去的经验(如类似结构化合物的分离)和/或用文献资料补充现有信息而另一种模式则直接开始色谱分离,而对样品的性质不大注意这两种HPLC的方法建立模式可分别称为理沦型与经验型初始分离一旦开始,可以根据类似的思路(理论的与经验的)选择进一步的实验。
2.分离的目的HPLC分离的目的必须十分明确,下面的问题在建立方法之初就应确定:(1)主要目的是什么?定量或定性,还是定性、定量同时做?;(2)是否有必要解析出样品的所有成分?譬如可能有必要分离出产品中的所有降解物或杂质,以使含量测定结果更加可靠,但却没必要将它们彼此完全分开。
(3)如要求定量分析,准确度与精密度需多大?样品主要成分的精密度通常能达到±1—2%,特别是不需样品预处理的情况。
(4)特殊化合物可能会以不同的样品形式出现(如:原料药,一种或多种形态,环保样品等)。
是否需要一种以上的HPLC方法?单一方法分离不同形态样品是否理想?(5)一次将分析多少样品?当必须同时处理大量样品时,运行时间将变得非常重要。
有时甚至为了缩短运行时间而以牺牲样品分离度作代价,如缩短柱长或加快流速。
当一次分析的样品数目超过10个,运行时间一般应控制在20min以内。
(6)将要使用该方法的实验室中,有哪些HPLC设备?色谱柱能否恒温系统能否做梯度洗脱?该方法是否可在不同设计与生产的设备上运行?方法建立实验开始之前,应明确对方法的这些要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
建立色谱分析方法需考查的内容
建立色谱分析方法除进行色谱系统适用性试验外,还必须进行方法学的考查,包括方法的精密度、准确度、检测限、定量限、线性与定量范围等。
有时还需要考查方法的专属性和耐用性。
1.精密度(precision)
是指在规定的测试条件下,同一个均匀样品,经多次进样测定所得结果之间的接近程度,反映了正常测定条件下分析方法的再现程度。
含量测定和杂质定量测定应考虑方法的精密度。
常用相对标准偏差来表示。
精密度分为方法精密度和仪器精密度。
重复性:在相同的条件下,由一个分析人员测定所得结果的精密度;
中间精密度:在同一个实验室,不同时间由不同分析人员用不同设备测定结果的精密度;重现性:在不同实验室由不同分析人员测定结果的精密度。
2.准确度(accuracy)
是指用该方法测定的结果与真实值或参考值接近的程度,是测量的系统误差和随机误差的综合,反映了方法所得结果对真值的接近程度。
回收率试验:在实际工作中经常用做回收率试验的方法来评价准确度,对已知标准品加入量的样品用待评定方法测定其含量作为回收量,计算回收量和加入量的百分数作为方法的回收率。
测定回收率的常用方法包括模拟样品法和标准加入法两种。
3.灵敏度(sensitivity)
又称响应值或应答值,一定浓度或一定量的样品进入色谱仪器(主要是检测器)后,进样量△Q与信号强度改变△R之比,称为仪器的灵敏度(S),即
S=△Q/△R
由于灵敏度只反映仪器对样品的敏感程度,而未考虑仪器的噪声对检测的影响,因此通常都用检测限来衡量。
4.检测限(detection limit)
是指试样中被测物质能被检测出的最低量,表示为能产生确证在样品中存在的被测组分信号所需要的该分析物的最低浓度或最小量。
一般表示为分析物质的浓度。
检测限是指在适当的置信概率被检出的组分最低浓度或最小量。
检测限的定义式:D=N/S N---仪器的噪声S---仪器的灵敏度
美国国家标准局,将检测下限分为三类:
(1) 仪器检测限:相对于背景仪器检测的最小的可靠信号,通常用信噪比(S/N )表示,
当S/N ≥3(或2)定义为仪器检测下限;
(2) 方法检测限:即某方法可检测的最小浓度。
通常用外推法可求得方法的检测下限。
其方法是在低浓度范围下测试3个浓度,每个浓度水平均分别测试多次计算其标准偏差,以标准偏差-浓度用线性回归法计算或绘制回归线,然后把回归线延长外推和纵坐标的交点的标准偏差即为方法的检测限,故这里是把浓度为零时的标准偏差定义为方法检测限。
(3) 样品检测限:即相对于空白可检测的最小样本含量。
定义样本检测限为3倍空白标
准偏差。
从统计学的观点 ,检测限应表示为样品信号为空白样品值加上3倍空白样品信号标准差对应的样品浓度,其数学表达式为:
D=A C σ
3⨯
σ――空白溶液10次以上测定的标准差;
C ――浓度接近检出限的样品液的实际浓度;
A ――测得的样品液的信号值。
式中A/c 应为校正曲线的斜率,表示被测组分的浓度或质量改变一个单位时校正曲线上
分析信号的变化量,即灵敏度。
5. 定量限(quantitation limit )
是指样品中被测物能被定量测定的最低量,其测定结果是定量分析应达到的准确度和精密度。
由于受到校正曲线在低浓度区域的非线性关系、试剂纯度、沾污等因素的影响,定量限一般都高于检测限,仪器方法定量限常用信噪比确定,以10倍于仪器噪声标准差时实际测得的浓度或质量表示。
6. 定量范围(range )
即一个定量分析方法的适用范围,指分析物浓度的上、下限范围。
在该定量范围内,应用本法能达到方法本身的精密度、准确度及线性等。
7. 线性(linearity )
是指在规定的范围内,实验结果和分析浓度成正比的能力。
在线性范围内可以直接应用分析方法建立的工作曲线或回归方程计算样品的浓度。