锐钛矿型TiO_2_10面本征点缺陷的理论研究
【国家自然科学基金】_锐钛矿型tio2_基金支持热词逐年推荐_【万方软件创新助手】_20140730
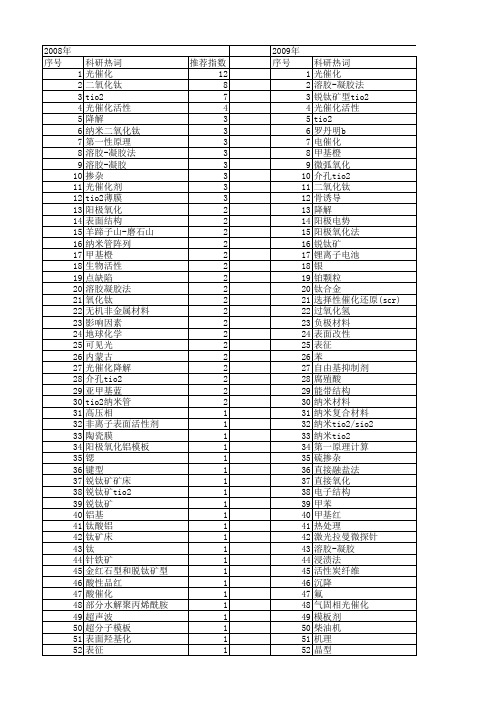
科研热词 光催化 溶胶-凝胶法 锐钛矿型tio2 光催化活性 tio2 罗丹明b 电催化 甲基橙 微弧氧化 介孔tio2 二氧化钛 骨诱导 降解 阳极电势 阳极氧化法 锐钛矿 锂离子电池 银 铂颗粒 钛合金 选择性催化还原(scr) 过氧化氢 负极材料 表面改性 表征 苯 自由基抑制剂 腐殖酸 能带结构 纳米材料 纳米复合材料 纳米tio2/sio2 纳米tio2 第一原理计算 硫掺杂 直接融盐法 直接氧化 电子结构 甲苯 甲基红 热处理 激光拉曼微探针 溶胶-凝胶 浸渍法 活性炭纤维 沉降 氟 气固相光催化 模板剂 柴油机 机理 晶型
科研热词 光催化 二氧化钛 tio2 光催化活性 降解 纳米二氧化钛 第一性原理 溶胶-凝胶法 溶胶-凝胶 掺杂 光催化剂 tio2薄膜 阳极氧化 表面结构 羊蹄子山-磨石山 纳米管阵列 甲基橙 生物活性 点缺陷 溶胶凝胶法 氧化钛 无机非金属材料 影响因素 地球化学 可见光 内蒙古 光催化降解 介孔tio2 亚甲基蓝 tio2纳米管 高压相 非离子表面活性剂 陶瓷膜 阳极氧化铝模板 锶 键型 锐钛矿矿床 锐钛矿tio2 锐钛矿 铝基 钛酸铝 钛矿床 钛 针铁矿 金红石型和脱钛矿型 酸性品红 酸催化 部分水解聚丙烯酰胺 超声波 超分子模板 表面羟基化 表征
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98
【国家自然科学基金】_光学能隙_基金支持热词逐年推荐_【万方软件创新助手】_20140802
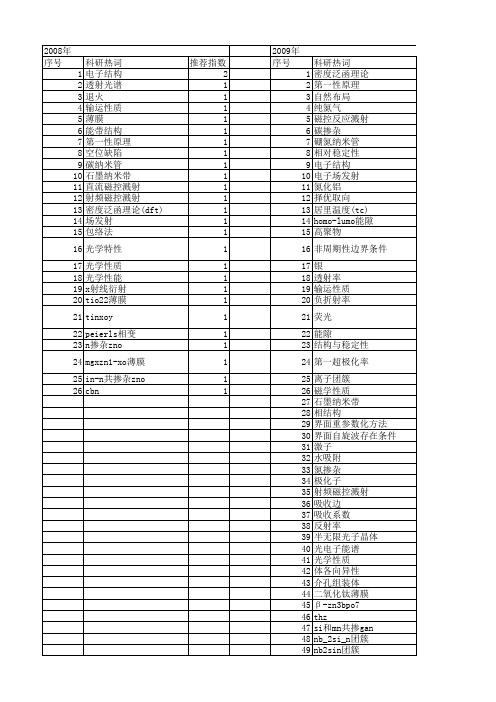
科研热词 密度泛函理论 第一性原理 自然布局 纯氮气 磁控反应溅射 碳掺杂 硼氮纳米管 相对稳定性 电子结构 电子场发射 氮化铝 择优取向 居里温度(tc) homo-lumo能隙 高聚物 非周期性边界条件 银 透射率 输运性质 负折射率 荧光 能隙 结构与稳定性 第一超极化率 离子团簇 磁学性质 石墨纳米带 相结构 界面重参数化方法 界面自旋波存在条件 激子 水吸附 氮掺杂 极化子 射频磁控溅射 吸收边 吸收系数 反射率 半无限光子晶体 光电子能谱 光学性质 体各向异性 介孔组装体 二氧化钛薄膜 β -zn3bpo7 thz si和mn共掺gan nb_2si_n团簇 nb2sin团簇 mg,zn,si,o和mn共掺gan mg cphf
2010年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43
科研热词 碳纳米管 电子结构 非线性光学 集合结构 锐钛矿型tio_2 色散关系 能隙重正化 能隙 第一超极化率 稀土离子发光 磁控反应溅射 硼氮共掺杂 电子吸收光谱 电子受体 电子供体 烷氧基噻吩 氮化铝 村底 杂环 晶格波 掺tm晶体 择优取向 射频磁控溅射 密度泛函理论(dft) 外场 噻唑生色分子 噁二唑 咔唑衍生物 吸附 单量子阱 半导体光学 前线分子轨道(fmo) 光谱 光致发光 光致亲水性能 光电功能材料 光学性质 光学双稳 光子雪崩 二阶非线性光学性质 二氧化钛薄膜 三氰呋喃 ce掺杂
2013年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
TiO_2光催化分解水制氢研究进展

TiO2光催化分解水制氢研究进展李国防1曹广秀1赵彦保21商丘师范学院化学系河南商丘4760002河南大学化学化工学院河南开封475004国家自然科学基金项目20671029资助2007208221收稿2008202229接受摘要综述了近几年改善TiO2光催化分解水制氢的方法措施。
向水中添加供电子物质可减少光生电子与空穴的复合添加碳酸盐或碘化物有利于光生电子与空穴分离TiO2表面沉积适量的金属颗粒也有利于实现电子和空穴分离但沉积太多的金属颗粒不但降低TiO2对光的吸收而且还可能成为光生电荷复合的中心掺杂合适的金属离子?ü 纬稍又誓芗犊砂裈iO2的吸光范围至拓宽可见光掺杂非金属元素使TiO2的带隙Eg变窄从而使TiO2的吸光红移更明显但掺杂离子有可能成为光生电荷复合的中心染料敏化或半导体复合有利于实现电荷分离提高光电转换效率。
将多种修饰方法有机结合起来制取氢是目前的一个研究方向最后分析了未来的研究重点。
关键词光催化制氢光催化剂修饰TiO2ProgressofPhotocatalyticWater2splittingUsingTiO2forHydrogenProductionLiGuofa ng1CaoGuangxiu1ZhaoYanbao21DepertermentofChemistryShangqiuNormalCollegeShangqiu4760002Coll egeofChemistryampChemicalEngineeringHenanUniversityKaifeng475004Abstract Theprogressofimprovementtechniquesinphotocatalyticwater2splittingusingTiO2forhydro genproductionisreviewed.Addingelectrondonorscanreducetherecombinationofphoto2gene ratedconductionbandelectronsandvalencebandholes.Additionofcarbonatesaltsoriodidecan enhancethephoto2excitedelectron∏holeseparation.Loadingofappropriatemet alparticlesont hesurfaceofTiO2canimprovephoto2excitedchargeseparation.Howeveranexcessofmetalpar ticlesmightnotonlyreducephotonabsorptionbyTiO2butalsobecomeelectron∏holerecombin ationcenters.AppropriatemetaliondopingonTiO2canexpanditsphoto2responsetovisibleregi onthroughformationofimpurityenergylevels.Aniondopingcausesthenarrowofthebandgapof TiO2andismoreeffectivethanmetaliondopingforredshift.Butdopedionstendtobecomerecom binationcenters.Dyesensitizationorsemiconductorcompositioncanresultinefficientchargese parationandimprovethephotocatalyticefficiency.Couplingdifferenttechniquesisoneofthedir ectionsofthefutureresearchforhydrogenproductionpotentialnewdirectionsrequiredinthisare aofresearcharehighlighted.Keywords PhotocatalysisHydrogenproductionPhotocatalystmodificationTiO2随着石油、煤炭等能源将会趋于枯竭寻找新的替代能源是世界各国关注的重点其中氢能被认为是一种最理想的绿色替代能源。
二氧化钛光催化原理

TiO?光催化氧化机理TiO?属于一种n型半导体材料,它的禁带宽度为3.2ev (锐钛矿),当它受到波长小于或等于387.5nm的光(紫外光)照射时,价带的电子就会获得光子的能量而越前至导带,形成光生电子(&);而价带中则相应地形成光生空穴(h+),如图1-1所示。
图IT Ti爲光电效应示意图Fi 此L-L Schematic diagram of phnto&lectric transfer effect on TiQ如果把分散在溶液中的每一颗TiO?粒子近似看成是小型短路的光电化学电池,则光电效应应产生的光生电子和空穴在电场的作用下分别迁移到TiO?表面不同的位置。
TiO?表面的光生电子e-易被水中溶解氧等氧化性物质所捕获,而空穴八则可氧化吸附于TiO?表面的有机物或先把吸附在TiO?表面的OHff 口9分子氧化成-0H1由基,• OH 自由基的氧化能力是水体中存在的氧化剂中最强的,能氧化水中绝大部分的有机物及无机污染物,将其矿化为无机小分子、反应过程如下:反应过程如下:CO和HO等无害物质。
+・1. . . 1. ■(3)h+eG热能h++OH-TOH⑸+ h + H 2O TOH + H +(6) e- +0 2T O2(7)O2+H+ THO2 ・⑻2H2OTO2+H 2O20)H2O2 +02 TOH + H +02(10)OH + dye T ・ T CO2 + H 2O(11)H+ dye T •—> C02 + H20(12)由机理反应可知,TiO?光催化降解有机物,实质上是一种自由基反应TiO 2光催化氧化的影响因素1 试剂的制备方法常用TiO 2光催化剂制备方法有溶胶一凝胶法、沉淀法、水解法等。
不同方法制得的TiO 2粉末的粒径不同,其光催化效果也不同。
同时在制备过程中有无复合,有无掺杂等对光降解也有影响。
TiO 2的制备方法在许多文献上都有详细的报道,这里就不再赘述。
TiO_2薄膜处理水中亚甲基蓝的动力学研究

化反应动力学方面做了一些工作u , 由于光催 但 化反应过程的复杂性, 不同研究者考虑的体系和对 象不一样 , 所得结果有差异. 深入研究光催化反应
k /  ̄ o 计 算得 到样 品的粒径为 3 .4n X ( cs ) 2 9 m.
第 2期
柳福提等 : i 薄膜处理水 中亚 甲基蓝的动力学研 究 TO
2 2 亚 甲基 蓝初始 浓 度对 降解 速 率 的影 响 亚 甲 .
l O~、 . 7 1 1 1×1 0~、 . 6 0 和 1 7 9X 1 ~ 1 4 2 X1 . 4 0
基 蓝的光催 化氧化 是 多相反 应 , 简化 动 力学 模 型 为 作 以下假设 :1 反 应发生 在气 一 、 一固相 的表 () 液 液 面 , 甲基 蓝的 降解 主要 是 ・ H 的作用 ;2 溶 液 亚 O ()
分光光度计 在最 大吸收波 长 6 5 n 6 m处测 定 溶液浓
本研 究 采 用 溶 胶 一凝 胶 法 制 备 纳 米 TO i 薄 膜, 并通过 X D对其 进行 了表征 , 到纳 米级 的活 R 得
度 的变化 , 光催化 降解率 叼按下式计 算 :
叩: × 0% ×10 , () 1
2 1 3月 00年 第3 3卷 第 2期
四川师范大学学报(自然科学 版)
Ju a o ScunN r M U ie t( a r Si c) or l f iha om nv ̄i N t  ̄ c ne n y u e
M a .. 01 r 2 0 V0l3 No 2 | 3. .
氧分压对TiO_2膜结构与光学性质的影响

(a1) 金红石的表面结构
(a2) 金红石的断面结构
(b1) 锐钛矿的表面结构
(b2) 锐钛矿的断面பைடு நூலகம்构
图 4 不同氧分压下两种晶体结构的 SEM 形貌图
响. (b) 较 (a) 因为只有一个晶向 ,可见晶体长得很 好 ,晶粒比较均匀 ,膜表面比较平滑. 2. 4 氧分压对 TiO2 膜光学性质的影响 2. 4. 1 折射率 n 和消光系数 k
朱 凤 赵 坤 赵 夔 王莉芳 全胜文
(北京大学重离子物理研究所超导腔组 , 北京 100871)
摘 要 :报道了用反应溅射法制备 TiO2 膜的实验研究. 详细研究了膜的沉积 、膜结构及其光学 性质 ,随溅射氧分压的变化. 随氧分压由 6 ×10 - 2 Pa 增加到 9 ×10 - 2 Pa 时 ,晶体结构由金红石 变到锐钛矿 ,氧分压超过 9 ×10 - 2 Pa 时趋向于无定形结构. 与膜结构密切相关的折射率 n 随 氧分压的增大由 2. 44 变到 1. 96 ,禁带宽度 Eg 则由大变小 ,然后再增大的变化 (3. 41 →3. 26 → 3. 42) . 关键词 : 反应溅射 ; TiO2 薄膜 ; 氧分压 ; 折射率 ; 禁带宽度 中图分类号 : O48 ; TQ028 文献标识码 : A
表 1 不同氧分压下 TiO2 膜的折射率 、消光
系数 、薄膜厚度与禁带宽度
氧分压
样品 编号
pO2 ×102
/ Pa
晶体 结构
折射率 消光系数
n
k ×103
(500 nm) (500 nm)
薄膜 厚度 d/ nm
禁带 宽度
Eg/ eV
N1 6. 9 金红石 2. 437 3 2. 133 413 3. 41
【国家自然科学基金】_锐钛矿型_基金支持热词逐年推荐_【万方软件创新助手】_20140730
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106
科研热词 光催化 溶胶-凝胶法 光催化活性 锐钛矿型tio2 二氧化钛 tio2 罗丹明b 纳米tio_2 电催化 甲基橙 水热法 机理 掺杂 微结构 微弧氧化 介孔tio2 骨诱导 降解 阳极电势 阳极氧化法 阳极氧化 锐钛矿 锂离子电池 银 铂颗粒 钛酸铋 钛合金 钒基锐钛矿型催化剂 选择性催化还原(scr) 选择性催化还原 过氧化氢 负极材料 表面改性 表征 苯酚 苯 自由基抑制剂 腐殖酸 能带结构 纳米管阵列 纳米材料 纳米晶岛 纳米复合材料 纳米压印 纳米tio2/sio2 纳米tio2 第一原理计算 稀磁半导体 硬脂酸法 硫掺杂 硅藻土 直接融盐法
机械激活 有机氯农药 有机染料 晶须 晶型 晶体结构 敏化 损伤 微弧氧化 形成机理 异构十三醇聚氧乙烯醚 密度泛函 室温铁磁性 实验条件 多弧离子镀 多孔微球 多孔二氧化钛微球 外场作用 复合材料 复合sno2/tio2 图形化表面tio2薄膜 固态反应 吸附相反应技术 合成粉体 可见光响应 反应离子刻蚀 反应气氛 协同效应 功能材料 制备 分布 分子机制 共掺杂 光解水 光电化学活性 光电化学 光学性能 光刻 光催化材料 催化性能 低红外发射率 低温燃烧合成 低温 介孔 二氧化钛薄膜电极 二氧化钛溶胶 二氧化钛(tio2) 乙二醇 主要地质特征 主元素和微量元素 α -pbo2型tio2 toc tioso4液 tio2纳米粒子
ZnO和ZnS本征点缺陷的理论研究
ZnO和ZnS本征点缺陷的理论研究马昌敏;刘廷禹;常秋香;罗国胤【摘要】基于第一性原理和热动力学方法,通过模拟计算分析了不同温度和分压下ZnS和ZnO晶体本征点缺陷的性质.振动熵的计算结果表明,在高温条件下,振动熵对缺陷形成能的贡献不能忽略.对比分析2种晶体本征点缺陷随环境条件变化的规律,结果表明,2种晶体的主导缺陷均为空位型.氧空位(V0)在ZnO中更易形成,富氧和低温条件有利用于ZnO的p型本征掺杂.而锌空位(Vzn)在ZnS中形成能最低,因此ZnS比ZnO更容易形成p型掺杂.研究还发现2种晶体的肖特基缺陷都不稳定,而弗伦克尔缺陷比较稳定.除ZnS反弗伦克尔缺陷外,有价态的缺陷对的形成能均比中性缺陷对的形成能低.【期刊名称】《高等学校化学学报》【年(卷),期】2016(037)005【总页数】6页(P932-937)【关键词】密度泛函理论;点缺陷;热力学;氧化锌;硫化锌【作者】马昌敏;刘廷禹;常秋香;罗国胤【作者单位】上海理工大学理学院,上海200093;上海理工大学理学院,上海200093;上海理工大学理学院,上海200093;上海理工大学理学院,上海200093【正文语种】中文【中图分类】O641ZnS和ZnO均为锌基Ⅱ-Ⅵ族化合物直接宽带隙半导体, 是非常重要的发光半导体材料, 均包含立方(闪锌矿)和六方(纤锌矿)2种晶体结构. ZnO通常以六方结构存在, 而ZnS通常为立方结构. ZnO的激子束缚能较大(≈60 meV), 透明度高, 广泛应用于发光二极管、荧光材料、光催化及太阳能光伏材料, 可变电阻及压电材料等领域[1~4]. ZnS则在阴极射线管的磷光体、电致发光器件、非线性光学器件、光催化剂及太阳能电池等方面有巨大的应用潜力[5]. 而实现ZnS和ZnO材料的n型和p型稳定掺杂, 是其在该领域应用的关键.ZnS和ZnO都具有单极性[6], 即: 如果n型掺杂容易, p型掺杂就非常困难, 反之亦然, 这成为其在光电领域应用的瓶颈, 目前已有很多利用共掺控制材料导电类型的报道[7~9]. 但要更好地控制导电类型, 本征点缺陷的深入研究尤为重要. 通过分析本征缺陷的形成来研究施主和受主中心的结构及物理机制, 从而更好地通过掺杂控制材料导电类型.有关ZnS和ZnO本征点缺陷的研究已有不少报道, 如王洪波等[10]研究了ZnO中主要点缺陷浓度与环境温度和氧分压的热力学关系. 徐彭寿等[11]研究了ZnO本征点缺陷的电子结构, 讨论了其对导电性的影响. 在富氧或富锌条件下, 对ZnO本征缺陷的研究也有报道[12,13]. Li和Deng 等[14]用第一性原理LDA, LDA+U方法对闪锌矿型ZnS的点缺陷进行了较深入的研究; Morozova 和Karetnikov等[15]研究了压强(100~200 MPa)和温度(900~1100 ℃)对ZnS的带隙和点缺陷平衡的影响. 关于ZnS高温电导和高温缺陷平衡的研究也有相关研究报道[16,17].上述研究都局限在特定温度(T)和氧分压或硫分压(pO2或pS2)条件下, 并未对ZnS 和ZnO晶体中点缺陷在整个T和pO2或pS2条件下进行全面系统的研究. 也未考虑振动熵对晶体缺陷形成能的影响, 因此, 本文提出在考虑振动熵对缺陷形成能贡献的情况下, 计算得到比较精确的缺陷形成能, 通过研究ZnS和ZnO晶体本征点缺陷的形成能随环境温度T, pO2或pS2和费米能级(EF)的变化关系, 获得外部条件对缺陷稳定性的影响, 为调控晶体n型和p型掺杂提供理论依据. 从而更好地通过掺杂控制材料导电类型.1.1 计算模型使用VASP软件和交换关联泛函[LDA+U(U=4.7 eV)][18,19], 对六方纤锌矿结构ZnO和立方闪锌矿结构ZnS晶体进行了模拟计算. 通过从96个原子的超晶胞中移走或增加原子来模拟缺陷, ZnO晶体结构如图1(A)所示.图1(A)中a(0.4167, 0.4444, 0.4413)为氧空位及氧被替位的位置, b(0.5833,0.5556, 0.5000)为锌空位及锌被替位的位置, c(0.5000, 0.3333, 0.5955)为填隙位置. ZnS晶体结构如图1(B)所示.图1(B)中a(0.5833, 0.6250, 0.3750)为硫空位及硫被替位的位置, b(0.6667,0.5000, 0.5000)为锌空位及锌被替位的位置, c(0.5000, 0.5000, 0.5000)为填隙位置. Zn的价电子结构为3d4s4p, S的价电子结构为3s3p, O的价电子结构为2s2p、ZnS和ZnO的平面波截断动能分别为360和400 eV, 布里渊区的积分采用2×2×2的Monkhorst-Pack k点取样求和. 经收敛测试k点和截断动能都已经达到收敛标准(能量差<0.001 eV). 结构优化收敛条件: (1) 每个原子的最大能量变化小于0.00001 eV; (2) 原子的最大位移小于0.0001 nm; (3) 每个原子上受到的最大作用力小于 0.5 eV/nm.1.2 缺陷形成能的计算点缺陷的吉布斯自由能是关于缺陷种类α、带电态q、硫(氧)分压p和温度T的函数, 表达式如下[20~22]:式中: Etotal(α, q)和Etotal(perfect)利用VASP计算获得, 分别代表电量为q的α缺陷的超晶胞经弛豫后的总能量和完整晶体的超晶胞经弛豫后的总能量; ni为从超晶胞中移除或添加α原子的数量, 如ni=nZn=1则是超晶胞中有一个Zn空位;μi(T, p)为通过VASP和热力学计算获得的对应原子随温度和分压变化的化学势;EVBM为完整晶体价带顶的能级; EF为指电子相对价带顶的费米能级; ΔV为含缺陷的超晶胞与完整超晶胞间的平均静电势之差; V0为晶体体积, 在计算中是一个常量. 本文用Janotti等描述的校正方式, 对缺陷形成能进行了校正, 具体过程见文献[18], 跃迁能级也进行了校正. 化学势μα(T,p)可以随温度和pS2(或pO2)的变化而变化. 根据Finnis等[23,24]的方法确定硫(氧)的化学势.本文用GULP软件[25]进行振动熵的计算. 该软件基于Born离子晶格模型, 采用基于核壳模型的半经验势方法计算. 用下式描述距离为r的2个离子间的相互作用: 式中:为库仑势,为Buckingham势, 其中A, ρ和C是短程势参数[26].利用GULP程序计算完整的和含不同孤立点缺陷晶体的振动熵, 计算公式为式中: Zvib为振动熵的配分函数.振动熵(TΔS)对缺陷形成能的贡献如图2所示. 计算结果表明, 缺陷的存在改变其周围原子的力场, 从而改变周围原子振动频率引起振动熵的改变, 只有反位硫的振动熵贡献随温度变化比较小, 在高温条件下振动熵贡献总体比较大, 不能忽视. ZnO与ZnS结果相似(此处不再列出).缺陷的形成能可很好地预测晶体的缺陷行为, 因此研究晶体缺陷态在不同环境条件(T, p, EF)下的缺陷形成能非常重要. 图3和图4分别为ZnS和ZnO在不同温度和分压条件下各种缺陷形成能随费米能级变化的关系图. 可以看出, ZnS和ZnO点缺陷的形成能随环境条件变化的基本规律是一致的, 施主型缺陷(VS, Zni, ZnS, VO, ZnO)的形成能是随着pO2或pS2的减少和温度的增加而减少, 但对于受主型缺陷(VZn, Si, SZn, Oi, OZn)变化规律是相反的. 替位型缺陷的形成能变化最快, 所有点缺陷的形成能对温度都比较敏感, 其中一个原因就是振动熵的贡献. 各点缺陷形成能随温度和分压的变化速率在2种晶体中相近, 在T=1300 K, 分压p从103 Pa到10-10 Pa变化时, ZnS中ZnS, VS和Zni缺陷形成能分别减少3.35, 1.62和1.63 eV, 而 Si, VZn和SZn分别增加1.68, 1.67和3.38 eV; ZnO中ZnO, VO和Zni缺陷形成能分别减少3.33, 1.64和1.69 eV, 而Oi, VZn和OZn分别增加1.69, 1.73和3.39 eV.在ZnO晶体中, 受主缺陷VZn在费米能级处于导带底附近有最低的形成能. 在温度不高的条件下. 在费米能级靠近价带顶区域, VO具有最低的形成能. 但不少研究结果表明, 氧空位的浓度不高[27,28]. 而本文的研究结果表明, 在高温低氧分压条件下, VO具有最低的形成能, 而Oi, OZn和VZn缺陷形成能非常高, 即受主缺陷浓度不高, 不会大量补偿氧空位提供的自由电子, 因此VO是ZnO晶体容易形成n型半导体的重要原因. 有许多研究[29]认为在富锌条件下, Zni缺陷是ZnO本征n型半导体的主要根源, 但是本文结果表明, Zni在所有的条件下都具有非常高的形成能, 它的浓度应该很低, 不能成为ZnO本征n型半导体主要根源.以往不少研究都认为ZnO是不能进行本征p型掺杂的[8,9]. 本征p型掺杂要求受主(VZn, Oi, OZn)有较低的跃迁能级和形成能, 而且施主缺陷不能对其进行完全有效补偿. VZn为浅受主缺陷, 在富氧低温条件下具有很低的形成能[图4(A)], 与此同时施主缺陷具有较高的形成能, 因此, 本文预测在富氧和低温条件下有利于ZnO的p型本征掺杂.在ZnS晶体中, 本征点缺陷形成能的变化规律与ZnO相似. ZnO晶体在一些特定条件下, 整个EF区域都只有VO的形成能最低, 也就是阴离子空位在ZnO中容易形成, 而ZnS晶体中大多数条件下VZn的形成能最低, 并且在ZnS中的VZn的跃迁能级更低, 因此ZnS比ZnO更容易进行本征p型掺杂, 这与富硫条件下ZnS应是弱p型半导体的结论[30]相一致.局部电中性是晶体稳定存在的基本要求, 因此, 带电点缺陷在晶体中是很难孤立存在的, 通常是以电中性的缺陷对形式存在, 因此, 研究弗仑克尔缺陷(阳离子空位与阳离子填隙形成电中性的缺陷对)、反弗仑克尔缺陷(阴离子空位与阴离子填隙形成电中性的缺陷对)和肖特基缺陷(阳离子空位与阴离子空位形成电中性的缺陷对)等缺陷对的形成能是很有必要, 计算公式如下:式中: Vα为硫空位或氧空位(VS或VO); αi为硫填隙或氧填隙(Si和Oi); μα为硫或氧的化学势(μS或μO).从图5可见, ZnS和ZnO肖特基缺陷的形成能都比弗伦克尔缺陷和反弗伦克尔缺陷高, 反弗伦克尔缺陷形成能又比弗伦克尔缺陷高, 所以2种晶体中肖特基缺陷都不稳定, 而弗伦克尔缺陷形成是比较稳定的. 对于肖特基缺陷和弗伦克尔缺陷, 由带电点缺陷组合成缺陷对的形成能比电中性的低, 如, 肖特基缺陷(V″Zn+V¨S)的形成能比)和)的形成能低, )的形成能比)的形成能低. 但是ZnS反弗伦克尔缺陷不同, )缺陷对的形成能比 + )的形成能更低[见图5(B)]. 这些关于缺陷对的结论与He等[31]报道的二氧化钛中的结论基本一致. ZnS和ZnO的缺陷对相比, ZnO的弗伦克尔缺陷和肖特基缺陷的形成能都比ZnS的高, 而ZnS的反弗伦克尔缺陷的形成能比ZnO高. 对3种缺陷对而言, 弗伦克尔缺陷各价态组合之间的能量差最大.综上所述, ZnS和ZnO 2种晶体的主导缺陷都是空位型. 施主型缺陷(VS, Zni, ZnS, VO, ZnO)的形成能是随着pO2或pS2的减少和T的增加而减少. 对于受主型缺陷(VZn, Si, SZn, Oi, OZn)变化规律是相反的, 并且替位型缺陷的形成能变化最快. 在ZnO中容易形成阴离子空位, 因此, ZnO中VO能够提供n型掺杂, 在富氧和低温条件下有利于ZnO的p型本征掺杂. 而ZnS中, 大多数条件下VZn的形成能最低, 因此ZnS比ZnO更容易进行本征p型掺杂. 阴离子空位(VS和VO)在高温, 低pO2或pS2条件下容易形成, 而阳离子空位VZn在富硫条件下容易形成. 2种晶体中肖特基缺陷都不稳定, 而弗伦克尔缺陷比较稳定, 并且除了ZnS反弗伦克尔缺陷以外, 由带电点缺陷组合成缺陷对的形成能比电中性的低. ZnO的弗伦克尔缺陷和肖特基缺陷的形成能都比ZnS的高, 而反弗伦克尔缺陷是ZnS比ZnO高. 对3种缺陷对而言, 弗伦克尔缺陷不同价态之间结合能量差异最大.† Supported b y the Foundation of Hujiang, China(No.B14004).【相关文献】[1] Triboulet R., Proc. SPIE, 2001, 4412, 1—8[2] Wang Z. L., J. Phys.: Condens. Matter., 2004, 16, 829—858[3] Ling J., Cong R. M., Acta Chim. Sinica, 2008, 66(18), 2070—2074(凌剑, 丛日敏. 化学学报, 2008, 66(18), 2070—2074)[4] Song J. Z., He Y., Zhu D., Chen J., Pei C. L., Wang J. A., Acta Phys.-Chim. Sin., 2011,27(5), 1207—1213(宋继中, 贺英, 朱棣, 陈杰, 裴昌龙, 王均安. 物理化学学报, 2011, 27(5), 1207—1213)[5] Xin D. S., Shi J. X., Pang Q., Acta Scientiarum Naturalium Universitatis Sunyatseni, 2003, 42(6), 125—127(邢德松, 石建新, 庞起. 中山大学学报(自然科学版), 2003, 42(6), 125—127)[6] Yan Y. F., Li J. B., Wei S. H., Al-Jassim M. M., Phys. Rev. Lett., 2007, 98, 1—4[7] Deng B., Luo M., Dong H. N., Electronic Quality, 2012, 1, 8—10(邓博, 罗敏, 董会宁. 电子质量, 2012, 1, 8—10)[8] Chen L. J., Li W. X., Dai J. F., Wang Q., Acta Phys. Sin., 2014, 63(19), 196101(陈立晶, 李维学, 戴剑锋, 王青. 物理学报, 2014, 63(19), 196101)[9] Yang T. Y., Kong C. Y., Ruan H. B., Qin G. P., Li W. J., Liang W. W., Meng X. D., Zhao Y.H., Fang L., Cui Y. T., Acta Phys. Sin., 2012, 61, 168101(杨天勇, 孔春阳, 阮海波, 秦国平, 李万俊, 梁薇薇, 孟祥丹, 赵永红, 方亮, 崔玉婷. 物理学报, 2012, 61, 168101)[10] Wang H. B., Zhang J. W., Yang X. D., Liu Z. L., Xu Q. A., Hou X., Acta Phys. Sin., 2005, 54(6), 2893—2898(王洪波, 张景文, 杨晓东, 刘振玲, 徐庆安, 侯洵. 物理学报, 2005, 54(6), 2893—2898)[11] Xu P. S., Shun Y. M., Shi C. S., Xu F. Q., Pang H. B., Science China A, 2001, 31(4), 358—365(徐彭寿, 孙玉明, 施朝淑, 徐法强, 潘海斌. 中国科学, A辑, 2001, 31(4), 358—365)[12] Kohan A. F., Ceder G., Morgan D., Phys. Rev. B, 1995, 61, 15019—15027[13] Oba F., Choi M., Togo A., Tanaka I., Sci. Technol. Adv. Mater., 2011, 12, 034302[14] Li P., Deng S. H., Zhang L., Liu G. H., Yu J. Y., Chem. Phys. Lett., 2012, 531, 75—79[15] Morozova N. K., Karetnikov I. A., Golub K. V., Gavrishchuk E. M., Yashina E. V., Plotnichenko V. G., Galstyan V. G., Inorg. Mater., 2004, 40(11), 1138—1145[16] Lott K., Shinkarenko S., Turn L., Nirk T., Opik A., Kallavus U., Gorokhova E., GrebennikA., Vishnjakov A., Phys. B, 2009, 404, 5006—5008[17] Lott K., Turn L., Volobujeva O., Leskela M., Phys. B, 2001, 949, 308—310[18] Anderson J., Van de Walle C. G., Phys. Rev. B, 2007, 76, 165202[19] Li P., Deng S. H., Zhang L., Yu J., Chem. Phys. Lett., 2012, 531, 75—79[20] Kohan A. F., Ceder G., Morgan D., Van de Walle C. G., Phys. Rev. B, 2000, 61, 15019[21] Van de Walle C. G., Neugebauer J., J. Appl. Phys., 2004, 95, 3851—3879[22] Zhang S. B., Northrup S. B., Phys. Rev. B, 1991, 67(17), 2339—2346[23] Batyrev I. G., Alavi A., Finnis M. W., Phys. Rev. B, 2000, 62(7), 4698—4707[24] Finnis M. W., Lozovoi A. Y., Alavi A., Ann. Rev. Mat. Res., 2005, 35, 167—179[25] Gale J. D., Chem. J. Soc. Faraday Trans., 1997, 93, 629—637[26] Oba F., Adachi H., J. Mater. Res., 2000, 15(10), 2168—2175[27] Janotti A., Van de Walle C. G., J. Cryst. Growth, 2006, 287, 58—65[28] Santana J. A., Krogel J. T., Kim J., Paul R. C. K., Reboredo F. A., Cond. Mat. Mtrl. Sci., 2014, 12, 23—30[29] Zhang S. B., Wei S. H., Zunger A., Phys. Rev. B, 2001, 63, 075205[30] Gai Y., Li J., Yao B., Xia J. B., J. Appl. Phys., 2009, 105, 113704[31] He J., Behera R. K., Finnis M. W., Acta Mat., 2007, 55, 4325—4337。
含铋掺杂TiO_2及其铋系光催化剂的研究现状
过 u — i漫反射测定 了 B3 TO 纳米粉体 的光吸 V Vs i i + - 收特 陛,结果显示该纳米粉体对 4 0m以上光有响 0n 应。 分析表 明 B 代 替 晶格 中的 T4形成 T— — i i + iO B 键 使得带 隙变窄 。wu等 用 溶胶 一凝胶 和表 面浸 渍 采
焦圣兵等 : 含铋掺杂 TO2 iBiblioteka 及其铋 系光催化 剂的研 究现状
照射下 , 有机 物取得 了 良好 的光 催化 降解 效果 。 i B 掺
分 析可知 B,C — i 催化 剂带 隙为 30V, iS N TO 光 . 吸收 e
杂浓度为 0 %时光生 电子和空穴分离效率最高。通 . 7
边为 4 5m。在紫外光和可见光下降解 R B和酚均 1n h
的研 究 现 状 进 行 了综 述 , 展 望 了 该类 可见 光 催 化 剂 的 发 展 前景 。 并 关 键 词 : ; 催 化 ; 催 化 剂 铋 光 光 中 图分 类 号 : 6 5 T 4 6 0 4 . Q 2. 4 6 文献标识码 : A
Re e r h o o t i o e O2 n imu h b s d p o o a ls s s a c fc n a n Bi p d Ti d b s t - a e h t c t y t d a a
为 B— iO作为 电子 捕 获剂 阻止 了电子 一空穴 对 的再
收稿 日期 :0 0 l— 5 2 1 一 12 作者 简介 : 圣兵( 9 6 )男 , 西 山阴人 , 理工程师 , 焦 1 7 一 , nI 助 毕业于太 原理工大学 , 本科学历 , 现从事地 球化 学样品分析研究工
Tio2材料的性质及应用
2.当氧的分压较低(如PO2 ≤5066.25Pa),底物S的浓度较高 (大于10-3mol/dm-3)时。温度效应取决于温度对有机底物和氧 吸附性能的影响
氮掺杂的二氧化钛带隙结构
表面光敏化 光敏化的作用机理
一
S*
hv
S
CB
A
S
CB A
VB 一
敏化剂激发后电子转移 CB
A
S
VB 催化剂再生
VB 电子转移给受体
表面还原处理
一方面,随着TiO2表面Ti3+位的增多,TiO2的费米能级 升高,界面势垒增大,减少了电子在表面的积累及与 空穴的进一步复合
其他影响因素 除了前面提过的影响因素外,外加氧化剂、光源、
光强、反应液中的盐等外界条件都可以对TiO2的光 催化活性产生一定的影响。
提高TiO2光催化活性的途径
目前的TiO2光催化剂存在两个问题:
①量子效率低 ②太阳能利用率低
解决方法:
贵金属沉积 复合半导体 离子掺杂修饰 表面光敏化 表面还原处理 表面鳌合及衍生作用 超强酸化
半导体电荷迁移速率增加,电子与空穴的 复合几率降低
活性增大
溶液pH值的影响
TiO2在水中的零电点(电荷为零的点)为pH=6.25
当溶液pH值较低时,TiO2表面质子化,带正电 荷,有利于光生电子向表面迁移
当溶液pH值较高时,由于OH-的存在,TiO2表 面带负电荷,有利于光生空穴向表面迁移
温度的影响
导带
Ec Ed
Ev
价带
N型半导体的能级
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
该表面存在比 率为 7 %—10 %的 空位 , 这些点 缺陷 会显著改变了材料表面的物理化学性质[ 13 ,14] .与金 红石 TiO2(110)面比较 , 可以期待的是在超高真空的 环境下退火处理 , 锐钛矿型 TiO 2(101)面表面低配位 的氧原子容易离开晶格位置而形成点缺陷 .但实验 已经发现低配位的氧原子形成的空位并不是唯一种 类的点缺陷 , 并且这些点缺陷的浓度可能小于金红 石 TiO2 (110)面[ 15] .由 于获得符合实验要求的 锐钛 矿型 TiO2 清洁表面十分困难 , 以及实验技术本身的 限制很难指出缺陷的类型[ 16] , 导致目前对锐钛矿型 TiO2 表面的点缺陷研究很少见报道 .运用可靠的理 论计算研究晶体表面 , 可以提供实验分析技术所不 能提供的数据 .平面波超软赝势方法已经被我们成 功地用 于 研究 材 料的 表 面的 原子 结 构 及电 子 结 构[ 4 , 5] , 本文将采用同样的计算方法研究 锐钛矿型 TiO2(101)面点缺陷的形成能以及几何结构 .
×3 进行分格 .后面的所有的计算均采用了相同的 设置 .本 文 所 有 的 计算 工 作 采 用 CASTEP[ 24] 软件 完成 .
定超胞常数的方法 , 仅仅让表面上的 12 层原子层进 行弛豫优化 .
表 1 几种计算方案分别进行 TiO2 几何结构参数 优化所得到的结果与实验值的比较
方案 PBE RPBE PW91 实验值[ 23]
5期
马新国等 :锐钛矿型 TiO2(101)面本征点缺陷的理论研究
31 21
原子几何结构及电子结构[ 4, 5 , 17—19] .交换关联能采用 广义梯度近似(GGA)中 PW-91 方案[ 20] .从表 1 中可
以看 出 , 该方 案 计 算 出 的 TiO2 几 何 结 构 参 数比 PBE[ 21] 和 RPBE[ 22] 方案更接 近实验值[ 23] .计算中平
本文工作是在文献[ 4 , 5] 中锐钛矿型 TiO2(101) 表面模型的基础上考虑表面点缺陷 , 即采用 slab 模 型模 拟 半 无 穷 大 晶 体(表 面).选 取 真空 厚 度 为 0.8 nm , 以保证上下两层表面之间的作用可以忽略 , 表面厚度选取 18 层原子 , 其完整表面能收敛度小于 0.02 J m2 [ 4] .锐钛矿型 TiO2(101)面最表层应终止于 两配位的 O 原子 , 次层为五配位的 Ti 原子 .考虑到 实际 TiO2 表面存在的空位和间隙原子的浓度可能 比较低 , 为此沿[ 010] 方向建立 2 ×2 表面超胞 , 降低 点缺陷的浓度 , 以减小相邻点缺陷间的相互作用 .用 该表面超胞模拟有缺陷的表面是足够的[ 25] .本文模 型中的点缺陷是通过在表面上移去或增加原子(Ti 或 O)的方式获得 , 分别考虑了 O 空位(Vo ), Ti 空位 (VTi), O 间隙(O i ), Ti 间隙(Ti i )以及不同位置的缺 陷结构 , 如图 1 所示 .为了节约计算时间 , 采用了固
VTi1 -0.1330 (-0.08) 0.0021 (-0.65) 0.0080 (-0.63) -0.0021 (-0.32) —
— -0.0170 (1.41)
对 O 间隙缺陷结构进行研究 , 发现 O 间隙原子 与周围的 O 原子形成了双键(键长为 0.1423 nm), 即 O 间隙原子容易吸附在晶格上的 O 位上 , 并与 Ti 成 键 , 形成 Ti —O O 二聚物 , 这与文献[ 26] 中计算的 结果十分相近 .特别值得关注的是 Ti i2 间隙原子的 出现对结构的影响比较小 .结果显示了 Ti i2 间隙原
2.计算方法与物理模型
本文计算采用了基于密度泛函理论的平面波超 软赝势方法 , 该方法已被广泛用来研究体相和表面
*教育部新世纪优秀人才支持计划(批准号 :NCET-04-0702), 国家自然科学基金(批准号 :50771047)资助的课题 . E-mail :jiangjj @mail .hust .edu .cn
3.2.点缺陷形成能
与体相结构中点缺陷不同的是研究表面点缺陷
不仅要考虑点缺陷类型 , 还要考虑点缺陷在表面上
第 57 卷 第 5 期 2008 年 5 月 1000-3290 2008 57(05)3120-06
物 理 学 报
ACTA PHYSICA SINICA
Vol .57, No .5 ,M ay , 2008 c2008 Chin.Phys.Soc.
锐钛矿型 TiO2(101)面本征点缺陷的理论研究*
表面类型 Perfect
O1 0 (-0.59)
O2 0 (-0.73)
O3 0 (-0.70)
O4 0 (-0.64)
T i1
0
(1.37)
Ti 2
0
(1.29)
VO1
—
— -0.0009 (-0.74) 0.0227 (-0.69) 0.0140 (-0.68) -0.0142 (1.23) -0.0255 (1.15)
不同类型的表面点缺陷对周围原子间的作用是 不同的 , 这些缺陷会影响周围原子位置及周围电子 的分布 .表 2 列出了表面空位缺陷导致周围原子的 弛豫结果 .对于 VO1 空位缺陷 , 弛豫后的总能比未弛 豫时要低 0.57 eV ;空位周围的 Ti1 和 Ti2 原子向空 位外移动的位移较大 , 向表面里分别弛豫了 0.0142 和 0.0255 nm , 这主要源于空位上有效的正电荷与邻 近的阳离子之间的静电排斥作用 , 或者说 O1 空位的 产生使两个 Ti 原子之间失去了屏蔽它们之间排斥 作用的原子 .根据原子球内总电荷数和有效离子半 径 , 即所谓的 Shannon Prewitt 半径 , Ti4 +离子和 O2 -离 子的半径分别取为 0.061 和 0.140 nm , 计算了缺陷 周围原子的 Mulliken 净电荷布居数 .从表 2 可以看 到围绕着 O1 空位的两 个 Ti 原子净 布居数分 别为 1.23 和 1.15 e , 与产生缺陷前的布居数相比较 , 均增 加了 0.14 e 的电荷 , 其他 Ti 原子的电荷改变 小于 0.02 e , 这说明空位的产生导致了 Ti 离子周围局部 电子密度的增加 , Ti 与 O 的键连作用有所增强 .
3 12 2
物 理 学 报
57 卷
察这些原子的 Mulliken 电荷布居数可以发现 , 为了 达到静电平衡 , 原子之间的电荷重新分布 .O1 ,O4 原 子周围的电子显著减少 , O1 —O4 之间的共价特征明 显 , 而 Ti2 原子周围的电子减少 , 显然与它形成低配 位的几何构形有关 .而对 VTi2 空位结构进行优化 , 发
子与周 围的 最近 邻 的五 个 O 原 子之 间 的距 离在 0.1953 —0.2138 nm 之 间 , 主晶 格结 构没 有显 著变 化 , 因而该缺陷形成能很低 .然而 Tii1 间隙原子存在 晶格间隙中 , 由于其周围最近 邻原子均是 Ti , 其排 斥作用相当大 , 导致了表面结构显著的扭曲 , 其总能 比 Ti i2缺陷结构高 1.35 eV , 因此该位置的 Ti 间隙缺 陷结构是不稳定的 .
现其结构同 VTi1 空位结构相似 .其中 O1 原子向表面 里有 较 大 的 弛 豫 , 致 使 O1 与 O3 的 距 离 仅 仅 有 0.1273 nm .这些说明表面 Ti 原子空位缺陷的出现也 会导致表面 O 原子团聚成氧气分子 .
表 2 表面空位缺陷导致的周围原子的弛豫(沿[ 101] 方向的位移 , 向上弛豫为正 , 向下弛豫为负 ;单位 :nm)及空位周围各原子 Mulliken 净电荷布居数(单位电量)
与 VO1 空位缺陷周围的 Ti 原子移动情况相似 ,
VTi1 空位缺陷上有效的负电荷与周围 O 原子间的静 电排斥作 用导致周 围 O 原 子向空 位外移 动 , 其 中 O2 , O3 原 子 分 别 向 [ 010] 方 向 移 动 了 0.042 和 0.026 nm , 而 O1 原子 由于失去 Ti1 的键 连作用 , 使 Ti2 —O4 达到 0.2979 nm , 而 O1 向 表面里 弛豫了 近 0.1 nm , O1 有填补 Ti 空位的趋势 .此时 O1 与 O 4 的 距 离 缩 短 至 0.1334 nm (氧 气 分 子 的 键 长 0.1214 nm), 这说明了还原气氛下失去 Ti1 原子 , 导 致表面 O 原子有自发地团聚成氧气分子的趋势 .考
马新国 江建军 梁 培
(华中科技大学电子科学与技术系 , 武汉 430074) (2007 年 6 月 13 日收到 ;2007 年 9 月 13 日收到修改稿)
采用平面波超软赝势方法计算了锐钛矿型 TiO2(101)面存在本征空 位和间隙点 缺陷的几 何结构以及 缺陷形成 能 .首先分析了点缺陷对表面结构的影响 , 发现不同类型缺陷导致缺陷周围 原子有不 同的位移趋 势 :O 空 位的产生 导致空位周围的 Ti 原子向空位外移动 , Ti1 和 Ti2 空位的产 生均使 O1 自发地 与周围的 O 原子团 聚 , Oi 原子 易被周 围的氧原子吸附而成键 , 而 Tii2 缺陷几乎对晶格结构没 有产生 影响 .对 TiO2(101)面 上可能出 现的几 种点缺 陷的形 成能进行了计算 , 结果表明 :在还原性气氛 下 , 虽然 Tii2 和 VO1 缺陷 均容 易出 现 , 但 Tii2 缺陷 的形 成能 比 VO1 缺 陷更 低 ;而在氧化性气氛下 , 表面的 Oi 和 VTi1 缺陷较容易出现 .最后 , 为了比较各种 缺陷结构的稳定性 , 还计算 了几种典 型表面缺陷的形成焓 .
关键词 :第一性原理 , TiO2 , 点缺陷 , 表面结构 PACC :6820 , 6170B