分子对接 相互作用力 discovery studio
“Discovery Studio 2.5中基于药效团的药物设计方法和应用 ”

36 Compounds
1150compounds
+ Training set (16) Ligandfit
化合物库筛选
可以构建自己的化合物库
database
安装其他化合物库,CNPD,ACD等 DS中自带的化合物库
构建具有活性预测能力的药效团
• 寻找与化合物活性相关的药效团特性 • 构建的药效团模型具有活性预测功能
实例分析2(先导化合物优化)
Training set:
Test set
Pharmacophore modes
IC50=1.43
IC50=0.004
实例分析3
qualitative
Search 3D database
Dock(Ligandfit)
基于特性结构产生药效团
• 对于训练集要求:
- 输入的分子结构具有多样性
catalyst
Conformer Generation • FAST • BEST
Discovery studio
Conformer Generation • FAST • BEST • CAESAR • Systematic Search • Random Search • Boltzmann Jump
Common Feature Pharmacophore Generation HipHop(quanlitative)
- 化合物数目在2-32个,6个左右比较理想 - 只选用具有活性的分子
- 需要包含Principal和MaxOmitFeat性质
• 方法总结 - 找到一组化合物的公共药效特性
- 不需要SAR的信息
单击此处编辑母版标题样式
DS2.5中基于药效团的药物 单击此处编辑母版文本样式 设计方法和应用 第二级
生物分子间相互作用的计算和分析

生物分子间相互作用的计算和分析生物分子是生命体中不可或缺的组成部分,它们之间的相互作用决定了生命体的结构、功能和行为。
了解生物分子间的相互作用对于研究生物过程、发现新药物和设计新材料都具有重要意义。
随着计算机技术的不断发展,越来越多的计算化学方法被应用于生物分子间相互作用的计算和分析。
本文将介绍几种常用的计算化学方法,并讨论它们的优缺点及适用范围。
一、分子动力学模拟分子动力学模拟(Molecular Dynamics,MD)是一种计算生物分子间相互作用的方法,它可以模拟分子在一段时间内的运动过程,并得到分子的结构、构象和能量等信息。
分子动力学模拟所需要的输入是分子的几何结构和原子的物理化学参数,如原子间作用力场、电荷分布、溶剂作用力和温度等。
MD模拟可以用于研究分子的稳定性、构象变化和生物过程等,也可以用于设计新药物或优化现有药物的结构。
MD模拟的主要缺点是计算时间较长,而且需要高性能计算资源。
二、量子化学方法量子化学是一种计算化学方法,可以预测分子的电子结构和能量,进而推断分子之间的相互作用。
量子化学方法有很多种,如分子轨道法、密度泛函理论等。
这些方法都需要求解分子的薛定谔方程,计算精度较高,适用于小分子和分子间相互作用能较强的系统。
由于计算复杂度较高,所以量子化学方法通常只用于理论研究和计算机辅助药物设计等方面。
三、分子对接分子对接是一种计算化学方法,用于预测分子之间的相互作用和在多种分子中寻找合适的配位体结合。
分子对接的基本步骤包括构建配体库和靶标结构、对接计算和评价等。
分子对接计算的输出是分子对接的能量和结合模式,可用于理解靶标与配体的相互作用、发现新药物和优化分子设计等方面。
分子对接方法的缺点是需要结合实验验证和大量的计算资源,且误差较大。
四、分子机器学习分子机器学习是一种新兴的计算化学方法,利用机器学习算法对分子的结构、能量和动力学等信息进行分析,以预测分子的相互作用和性质。
分子机器学习基于大量的分子数据库和相关数据集,通过提取特征、学习和预测等步骤,可以快速准确地预测分子性质和相互作用。
共价结合分子对接

共价结合分子对接在现代化学和生物学的交叉领域,分子对接技术正发挥着越来越重要的作用。
其中,共价结合分子对接作为一种特殊的对接方式,为药物设计、材料科学等领域带来了新的思路和方法。
要理解共价结合分子对接,首先得明白什么是分子对接。
简单来说,分子对接就是在计算机模拟的环境中,让两个或多个分子相互靠近、匹配,预测它们之间可能形成的相互作用和结合模式。
就好像把两块拼图尝试拼在一起,看看哪一种组合方式最合适。
而共价结合则是一种特殊且相对更强的分子间相互作用方式。
在共价结合中,分子之间通过共享电子对形成稳定的化学键。
这种化学键的形成与传统的非共价相互作用(如氢键、范德华力等)相比,具有更高的稳定性和特异性。
那么,为什么我们要关注共价结合分子对接呢?这得从实际应用说起。
在药物研发领域,传统的药物大多通过非共价相互作用与靶点结合。
然而,这类药物可能存在结合亲和力不够强、作用时间不够持久等问题。
共价结合的药物则有可能克服这些不足。
通过精准地设计能够与靶点发生共价结合的药物分子,并利用分子对接技术预测其结合模式和效果,可以大大提高药物研发的成功率。
在共价结合分子对接中,关键的挑战之一是准确地描述共价键的形成过程。
这需要对化学反应的机理有深入的理解,同时要能够在计算模型中合理地模拟化学反应的能量变化。
此外,由于共价结合通常涉及到特定的官能团和反应活性位点,因此对分子结构的准确表征和分析也至关重要。
为了实现有效的共价结合分子对接,研究人员通常会采用一系列的技术和方法。
首先,需要建立高精度的分子力场,以便准确地计算分子之间的相互作用能。
这些力场不仅要考虑传统的非共价相互作用,还要能够准确地描述共价键的形成和断裂过程。
其次,利用量子化学计算方法可以对关键的化学反应步骤进行详细的研究,为分子对接提供更准确的能量信息。
在实际操作中,共价结合分子对接的流程一般包括以下几个步骤。
第一步是准备受体和配体的结构。
这需要从实验数据或者通过同源建模等方法获得受体的三维结构,同时对配体进行结构优化和预处理。
分子对接简要介绍
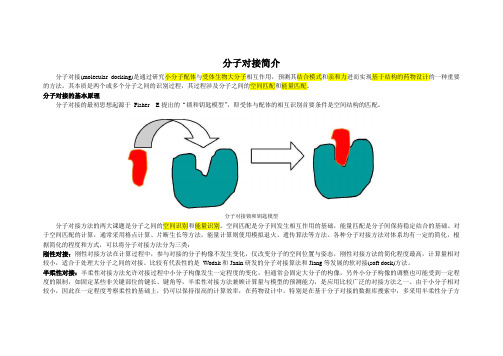
分子对接简介分子对接(molecular docking)是通过研究小分子配体与受体生物大分子相互作用,预测其结合模式和亲和力进而实现基于结构的药物设计的一种重要的方法。
其本质是两个或多个分子之间的识别过程,其过程涉及分子之间的空间匹配和能量匹配。
分子对接的基本原理分子对接的最初思想起源于Fisher E提出的“锁和钥匙模型”,即受体与配体的相互识别首要条件是空间结构的匹配。
分子对接锁和钥匙模型分子对接方法的两大课题是分子之间的空间识别和能量识别。
空间匹配是分子间发生相互作用的基础,能量匹配是分子间保持稳定结合的基础。
对于空间匹配的计算,通常采用格点计算、片断生长等方法,能量计算则使用模拟退火、遗传算法等方法。
各种分子对接方法对体系均有一定的简化,根据简化的程度和方式,可以将分子对接方法分为三类:刚性对接:刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,刚性对接方法的简化程度最高,计算量相对较小,适合于处理大分子之间的对接。
比较有代表性的是Wodak和Janin研发的分子对接算法和Jiang等发展的软对接(soft dock)方法。
半柔性对接:半柔性对接方法允许对接过程中小分子构像发生一定程度的变化,但通常会固定大分子的构像,另外小分子构像的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等,半柔性对接方法兼顾计算量与模型的预测能力,是应用比较广泛的对接方法之一。
由于小分子相对较小,因此在一定程度考察柔性的基础上,仍可以保持很高的计算效率,在药物设计中,特别是在基于分子对接的数据库搜索中,多采用半柔性分子方法。
其代表性软件是DOCK和AutoDock。
柔性对接:柔性对接方法在对接过程中允许研究体系的构像发生自由变化,由于变量随着体系的原子数呈几何级数增长,因此柔性对接方法的计算量非常大,消耗计算机时很多,适合精确考察分子间识别情况。
其中比较有代表性的方法有Accelrys 公司发展的基于分子力学和分子动力学的分子对接方法及Affinity 软件。
小分子对接

小分子对接一、小分子对接的概念1.1 什么是小分子对接小分子对接是一种计算化学方法,用于预测小分子与目标蛋白之间的结合方式和亲和力。
通过模拟和评估不同构象的小分子与蛋白的相互作用,可以为药物研发提供有价值的信息。
1.2 小分子对接的应用小分子对接广泛应用于药物设计和发现领域。
它可以帮助研究人员筛选候选化合物、优化药物分子、预测蛋白结合位点以及了解药物与靶点之间的相互作用机制。
二、小分子对接的方法2.1 理论基础小分子对接的方法基于分子力学和量子力学的原理。
它考虑了靶标蛋白和小分子之间的非共价相互作用,包括范德华力、氢键、离子键等。
通过计算这些相互作用的强度和方向性,可以预测小分子与蛋白结合的方式。
2.2 对接软件目前,市场上有许多不同的小分子对接软件可供选择。
其中一些软件采用基于力场的方法,如AutoDock和DOCK,而另一些则使用基于量子力学的方法,如GOLD和Schrodinger Suite。
每种软件都有其独特的算法和特点,研究人员可以根据实际需求选择合适的软件进行研究。
2.3 对接流程小分子对接通常包括以下几个步骤: 1. 靶标蛋白和小分子的准备:包括蛋白和小分子的结构优化、质子ation和离子化等。
2. 探索蛋白的活性位点:确定蛋白上可能与小分子结合的活性位点,这通常通过分子对接软件中的搜索算法实现。
3. 对接搜索:在活性位点附近对小分子进行搜索,尝试找到最佳的结合构象。
4. 结合模式评估:评估不同构象的亲和力和稳定性,选择最佳的结合模式。
5. 结果分析和优化:分析对接结果,优化小分子的化学性质以及与蛋白的相互作用。
三、小分子对接的挑战和展望3.1 挑战小分子对接面临着许多挑战,包括: - 结构灵活性:蛋白和小分子都存在结构的灵活性,如构象变化和溶剂效应等,这增加了对接的复杂性。
- 蛋白靶点的多样性:不同蛋白靶点之间的结构和功能差异很大,因此需要针对不同靶点进行优化和调整。
分子对接结果判断

分子对接结果判断分子对接(molecular docking)是一种计算化学方法,用于研究药物分子与靶标分子之间的相互作用。
通过模拟分子间的相互作用,分子对接可以评估药物与靶标的结合能力,并预测它们之间的结合模式。
在药物研发和生物化学研究中,分子对接是一个重要的技术手段。
本文将解释分子对接的基本原理,并讨论如何评估和判断分子对接的结果。
一、分子对接的基本原理分子对接的过程可以分为三个主要步骤:准备阶段、对接计算和结果评估。
首先,需要对药物和靶标分子进行准备。
这包括优化药物和靶标分子的几何构型,并预测它们的电荷分布。
接下来,通过对接计算,模拟药物和靶标之间的相互作用。
常见的对接算法包括基于引力亲和力场的对接方法和基于机器学习的对接方法。
最后,根据分子对接的计算结果,评估药物和靶标的结合能力和结合模式。
二、评估分子对接结果的指标为了准确评估分子对接的结果,我们需要考虑多个指标。
以下是常用的评估指标:1. 结合能(binding energy):结合能是指药物与靶标分子结合时释放或吸收的能量。
结合能越低,表示药物与靶标结合越稳定。
因此,负的结合能值通常表示较好的结合。
2. 位点亲和力(affinity):位点亲和力反映了药物与靶标结合时在位点上形成的非共价相互作用。
位点亲和力越高,表示药物与靶标结合得越紧密。
3. 相互作用模式(interaction pattern):相互作用模式描述了药物与靶标之间的结合方式。
通过观察氢键、疏水作用等相互作用,可以判断药物与靶标的结合模式是否合理。
三、判断分子对接结果的方法为了准确判断分子对接的结果,我们可以采用以下方法:1. 验证实验:将计算得到的分子对接结果与实验数据进行对比。
如果计算结果与实验结果一致,则说明分子对接的结果是可靠的。
2. 分析分子结合模式:通过观察药物与靶标之间的相互作用模式,判断药物是否与靶标结合得紧密且稳定。
如果药物与靶标之间存在合理的相互作用,说明分子对接结果可信。
分子对接的原理及应用
分子对接的原理及应用分子对接是一种计算化学方法,用于预测分子之间的结合方式和强度。
其主要原理是通过计算机模拟将配体分子(如药物分子)与受体分子(如蛋白质)进行匹配,以预测它们之间的相互作用。
分子对接在药物研发、酶机制研究和生物科学领域有着广泛的应用。
分子对接的具体过程包括两个主要步骤:搜索和评分。
在搜索阶段,计算机会生成一系列可能的配体构象,并将其与受体分子进行匹配。
搜索算法可以是基于力场的方法,如分子力学模拟、分子动力学模拟等,也可以是基于聚类、遗传算法等优化方法。
评分阶段则是根据一定的评分函数,对每个匹配构象进行打分,并选出具有最高得分的构象作为最佳配体-受体结合模式。
分子对接方法在药物研发中的应用非常广泛。
首先,分子对接可以用于筛选和优化潜在药物分子。
通过计算机模拟,可以预测不同小分子化合物与目标蛋白的结合方式和强度,从而筛选出具有潜在生物活性的化合物。
此外,对已有药物分子库进行虚拟筛选,可以加速药物发现的进程,减少实验成本和时间。
其次,分子对接方法可以用于解析药物-受体结合机制。
通过分析配体与蛋白质结合的方式和作用力,可以推断出配体与受体之间的相互作用方式和键合性质,为药物设计和优化提供理论指导。
这对于理解蛋白质的功能和构象变化,以及针对特定靶标的药物策略的制定都具有重要意义。
此外,分子对接技术也在计算机辅助药物设计中发挥了重要作用。
通过结合分子对接和分子动力学模拟等方法,可以优化药物的构象、增强药物-受体的稳定性,提高药物的选择性和亲和性。
相较于传统的药物设计方法,计算机辅助药物设计能够快速生成有生物活性的小分子候选物,同时在设计药物的特性和活性方面也更加灵活和精确。
不仅在药物研发中,分子对接方法也被广泛用于酶机制研究和生物科学领域。
在酶机制研究中,分子对接可以模拟底物和酶的结合,揭示酶的结构和功能,并推断催化机制。
在生物科学领域,分子对接还可以用于预测蛋白质的结构和功能,以及蛋白质和其他生物分子的相互作用,有助于解析生命过程中的分子机制。
Discovery Studio3.0中基于药效团的药物设计方法和应用讲义-2011年3月21日
矿产资源开发利用方案编写内容要求及审查大纲
矿产资源开发利用方案编写内容要求及《矿产资源开发利用方案》审查大纲一、概述
㈠矿区位置、隶属关系和企业性质。
如为改扩建矿山, 应说明矿山现状、
特点及存在的主要问题。
㈡编制依据
(1简述项目前期工作进展情况及与有关方面对项目的意向性协议情况。
(2 列出开发利用方案编制所依据的主要基础性资料的名称。
如经储量管理部门认定的矿区地质勘探报告、选矿试验报告、加工利用试验报告、工程地质初评资料、矿区水文资料和供水资料等。
对改、扩建矿山应有生产实际资料, 如矿山总平面现状图、矿床开拓系统图、采场现状图和主要采选设备清单等。
二、矿产品需求现状和预测
㈠该矿产在国内需求情况和市场供应情况
1、矿产品现状及加工利用趋向。
2、国内近、远期的需求量及主要销向预测。
㈡产品价格分析
1、国内矿产品价格现状。
2、矿产品价格稳定性及变化趋势。
三、矿产资源概况
㈠矿区总体概况
1、矿区总体规划情况。
2、矿区矿产资源概况。
3、该设计与矿区总体开发的关系。
㈡该设计项目的资源概况
1、矿床地质及构造特征。
2、矿床开采技术条件及水文地质条件。
Discovery Studio官方教程--创建2D QASR模型
构建二维定量构效关系模型(2D-QSAR )教程介绍药物设计中,当受体的结构未知时,基于结构的药物设计方法(主要为分子对接)将无计可施。
而QSAR 方法是基于小分子配体的药物设计方法,目的是采用数理统计方法研究和揭示化合物活性与其分子结构或物理化学特征之间的定量变化规律。
如果能够搜集到一系列结构类似物的生物活性数据,则可以采用QSAR (定量构效关系)的方法预测未知化合物的相关活性。
DS-QSAR 中包括以下几种产生模型的方式:♦ 贝叶斯分类(Bayesian categorization )♦ 遗传函数逼近(Genetic Function Approximation ,GFA )♦ 多重线性回归(Multiple Linear Regression ,MLR )♦ 偏最小二乘法(Partial Least Squares ,PLS )♦ 递归分区(Recursive Partitioning ,RP )本教程采用2D-QSAR 的方法研究甘氨酸/N-甲基-D-天冬氨酸抗肿瘤抑制剂的二维定量构效关系。
数据来源为如下的参考文献:2-Dimensional Quantitative Structure-Activity Relationship Modeling Study of Glycine/N-methyl-D-aspartate Antagonist Inhibition: Genetic Function Approximation Vis-à-vis Multiple Linear Regression Methods ,Nitin S. Sapre, Nilanjana Pancholi, Swagata Gupta and Arun Sikarwar ,Acta Chim. Slov . 2007, 54, 797–804 。
参考文献中,作者采用GFA 和MLR 两种线性回归的方法构建构效关系模型。
5.7分子对接基本理论
药物设计学分子对接1.基本原理aa分子对接是直接药物设计的重要方法之一,其本质是两个或多个分子之间的识别过程,涉及分子之间的空间匹配和能量匹配;最初思想起源于“锁-钥模型”(Lock-and-key theory),即一把钥匙开一把锁。
分子对接计算就是将配体小分子放置于受体的活性位点,并寻找其合理的取向和构象,使得配体和受体的形状和相互作用的匹配最佳。
对接类型方法特点计算量应用刚性对接参与对接的分子构象不发生变化,仅改变分子的空间位置与姿态简化程度最高,计算量相对较小适合于大分子之间的对接半柔性对接允许对接过程中小分子构象发生一定程度的变化,但通常会固定大分子的构象,另外小分子构象的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等兼顾计算量与模型的预测能力应用比较广泛柔性对接允许研究体系的构象发生自由变化计算量非常大,消耗机时很多适合精确考察分子间的识别常用于分子对接的软件包括:CDOCKER(Discovery Studio)、GOLD、AutoDock、Glide(Maestro)等CDOCKER采用半柔性的对接方法,可以产生高精度的对接结果所需功能和模块:Discovery Studio Client/Macromolecules /Small Molecules /Receptor-Ligand Interactions实验内容使用DS中CDOCKER模块将多奈哌齐药物分子对接至其作用靶标乙酰胆碱酯酶的活性口袋中,找出两者结合的最佳构象状态,并分析相互作用。
所需数据文件:晶体复合物pdb文件(1EVE.pdb)——Acetylcholinesterase + Donepezil (Aricept)配体mol2文件(Donepezil.mol2)Ref.Kryger, G. et al., Structure Fold. Des.1999,7: 297-307谢谢。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
分子对接相互作用力discovery studio
分子对接是一种主要用于计算机辅助药物设计的方法,可用于模拟和预测药物与靶标分子之间的相互作用。
在分子对接中,一方是药物分子(ligand),另一方是靶标分子(receptor)。
这两个分子通过非共价相互作用力进行结合,形成稳定复合物。
因此,对于药物发现研究而言,了解药物和靶标分子之间相互作用力的特性至关重要。
Discovery Studio是一种用于计算机辅助药物设计和药物发现的软件平台。
它内置了多种用于分子对接的工具和算法,可以帮助研究人员进行药物分子的筛选和优化,提高研发效率。
本文将以Discovery Studio的分子对接工具为主题,介绍其在药物发现中的应用和相互作用力的计算方法。
一、Discovery Studio分子对接的原理及流程
在Discovery Studio中,分子对接的过程可以大致分为以下几个步骤:准备受体结构、准备配体结构、建立格点和计算能量。
1. 准备受体结构
受体结构一般是指药物的靶标蛋白,可以从真实蛋白结构数据库中获取。
在进行分子对接之前,需要对受体结构进行优化和准备工作。
这包括去除水分子、修复缺失的原子、填充缺失的氢原子,并对蛋白进行能量最小化的处理。
2. 准备配体结构
配体结构是指药物分子或其他小分子化合物。
首先,需要将配体结构进行优化和准备工作。
这包括去除水分子、修复缺失的原子、填充缺失的氢原子,并对配体进行能量最小化的处理。
此外,需要确定配体的荷电状态和形式,如药物分子的离子化状态。
3. 建立格点
在分子对接过程中,通常会建立一个三维网格(grid)作为搜索空间,用于计算配体与受体之间的相互作用力。
这个网格的构建需要选择合适的参数和方法。
一般而言,可以使用栅格框(grid box)来定义搜索空间的大小和位置。
4. 计算能量
在分子对接过程中,需要计算配体与受体之间的相互作用能量。
这些能量包括范德华力、库仑力、极化等效力等。
计算能量的方法有很多,比如基于物理模型的计算方法(如精细化力场),以及基于经验的计算方法(如部分忽略特异性位点势能的能量评分函数)。
二、相互作用力的计算方法
Discovery Studio中常用的相互作用力的计算方法主要包括范德华力、库仑力、氢键、疏水效应等。
1. 范德华力
范德华力是一种由分子之间的诱导偶极、诱导多极和位形相关的分子间作用力。
计算范德华力的方法有多种,如基于荷电层势(Coulomb field)和散射理论。
2. 库仑力
库仑力是由分子之间的静电相互作用力引起的。
它可以是吸引力(正负电荷之间)或排斥力(相同电荷之间)。
库仑力可以通过计算分子之间的电荷与距离之积得到。
3. 氢键
氢键是一种特殊的非共价相互作用力,可以形成在氢原子与电负原子(如氮、氧、氟)之间。
氢键的强度和方向性可以通过计算氢键键能和键长来评估。
4. 疏水效应
疏水效应是溶剂中非极性分子或基团受到的相互作用力。
它是一种驱动分子在水溶液中聚集或形成复合物的重要力量。
疏水能力可以通过计算分子的溶剂表面积(SASA)来评估。
三、Discovery Studio分子对接的应用
Discovery Studio的分子对接工具在药物发现和研究中具有广泛的应用。
它可以用于虚拟筛选、药物重设计、药效团预测等方面。
1. 虚拟筛选
虚拟筛选是使用计算机模拟方法从大量化合物中筛选出具备潜力的候选分子。
在
Discovery Studio中,可以利用分子对接工具对候选配体库进行快速和高效的筛选,将潜在的药物分子纳入进一步实验验证的范围。
2. 药物重设计
药物重设计是指通过对已有药物结构的优化和修改,获得具有更好活性和选择性的新药物分子。
在Discovery Studio中,可以进行结构基于药物设计和分子动力学模拟,优化药物分子的对接得分和性质,从而改善药物的效力和药代动力学。
3. 药效团预测
药效团是指若干能够与靶点相互作用的共同基团或空间构型。
在Discovery Studio中,可以利用分子对接工具预测药效团与靶标分子之间的相互作用,进而预测药物的活性和作用机制。
总结
在药物发现研究中,分子对接是一种重要的计算方法,可以帮助研究人员预测和优化药物与靶标分子之间的相互作用。
Discovery Studio作为一种专业的计算机辅助药物设计软件平台,具备多种分子对接工具和算法,可以帮助研究人员进行药物分子的筛选、优化和设计。
同时,Discovery Studio还提供了丰富的相互作用力的计算方法,如范德华力、库仑力、氢键、疏水效应等,可用于评估药物与靶标分子的相互作用特性。
相信随着计算机技术的进一步发展,分子对接和
Discovery Studio的应用将在药物发现研究中发挥更加重要的作用。