上市后临床跟踪管理系统程序

1.PURPOSE
The purpose of this work instruction is to define the process to determine and
document whether a post-market clinical follow-up study is required forTDI
Foot/Ankle Array 8ch medical devices bearing the CE mark. The process will lead
to a determination of whether a post-market clinical follow-up study is required and provide guidance for post-market clinical monitoring requirements if a study is not
required.
2.SCOPE
The work instruction applies to all medical device businesses and sites operating
under the TDI Foot/Ankle Array 8ch Healthcare Quality Management System.
Only medical devices bearing the CE Mark will be required to follow this work
instruction.
3.REFERENCES
3.1.External References
https://www.360docs.net/doc/f29354697.html,ws
?Council Directive 93/42/EEC of 14 June 1993 concerning medical devices including amendments through 05 September 2007
3.1.2.Guidance Documents
?European Commission Enterprise-Directorate-General MEDDEV 2.12-2 Guidelines on Post Market Clinical Follow-Up dated May 2004
?MEDDEV 2.7.1 Rev.3 guidelines on medical device-clinical evaluation-a guide for manufacturers and notified bodies dated April 2009
?GHTF Post-Market Clinical Follow-Up Studies; SG5(PD)N4R7 (Proposed document 23 July 2008)
?GHTF Clinical Investigations; SG5(PD)N3R7 (20 January 2008)
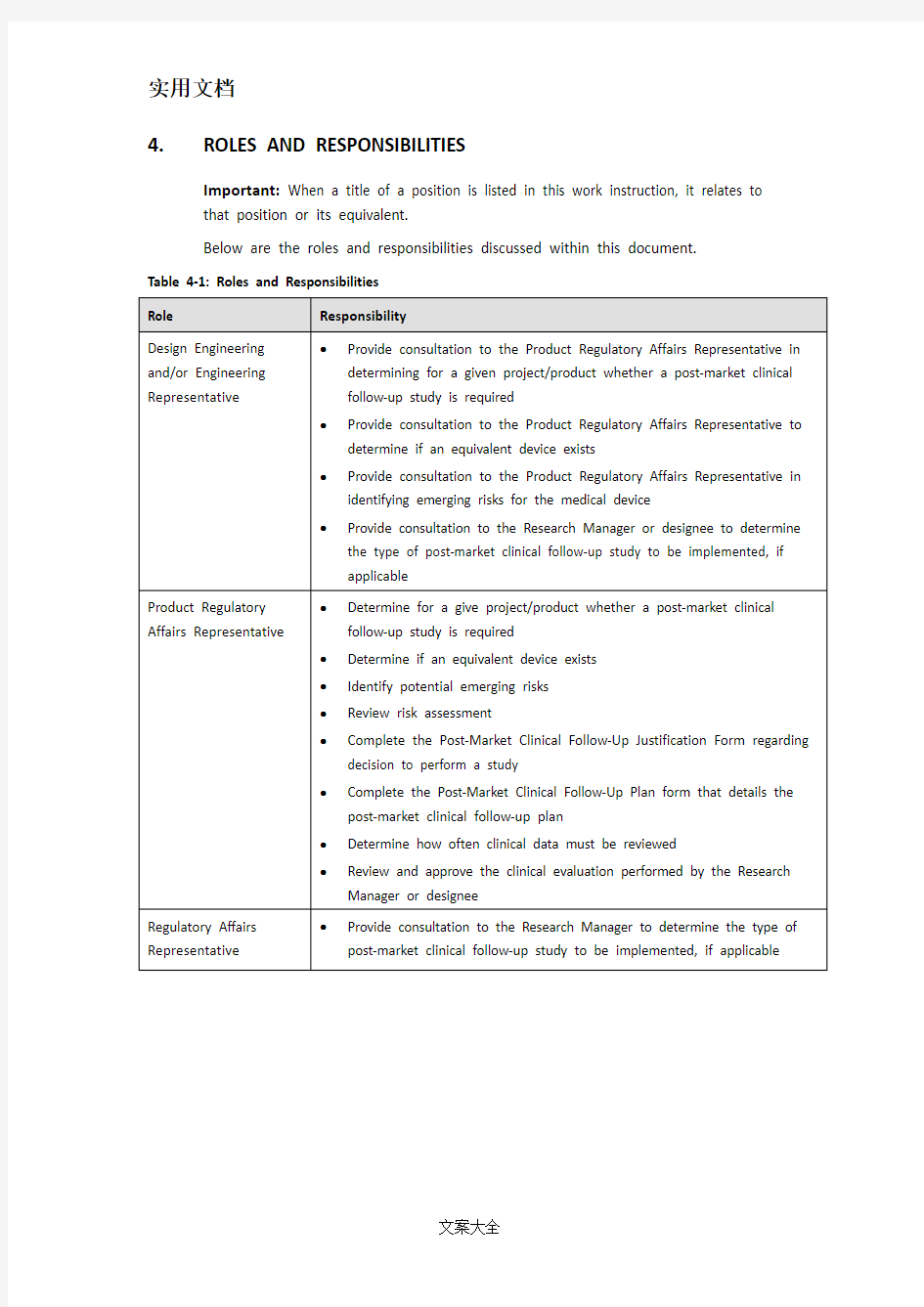
4.ROLES AND RESPONSIBILITIES
Important: When a title of a position is listed in this work instruction, it relates to that position or its equivalent.
Below are the roles and responsibilities discussed within this document.
Table 4-1: Roles and Responsibilities
Table 4-1: Roles and Responsibilities
5.WORK INSTRUCTION
Post-market clinical monitoring is an essential element in establishing long term
safety follow-up data and possible emergent risks for medical devices. These risks and data cannot adequately be detected and characterized by relying solely on
pre-market clinical investigations.
Post market clinical monitoring may include a combination of several strategies: ?Product complaint review
?Post-market event reporting review of users and patients
?Literature review
?Post-market clinical follow-up studies (PMCFS)
This work instruction was created to determine when a PMCFS is necessary to
maintain an adequate post-market surveillance system, as required by the Medical Device Directive 93/42/ECC (MDD) as amended by MDD 2007/47/EC. It will also
provide guidance on the post-market clinical monitoring requirements if a PMCFS is not required.
Figure 5-1: High-Level Process Overview for Post-Market Clinical Follow-Up
PMCFS
Determination
5.1.General Requirements
5.1.1.Prior to M3 sign-off, the Product Regulatory Affairs Representative in consultation
with the Research Manager or designee and the Design Engineering and/or
Engineering Representative shall determine for a given project/program whether a PMCFS is required. They shall also determine the post-market clinical follow-up
plan.
5.1.2. A PMCFS may not be required for products for which medium/long-term clinical
performance and safety is already known from previous use of the device or where other appropriate post-market surveillance activities would provide sufficient data to address the risks.
5.2.Determining the Type of Post-Market Clinical Follow-Up
Required
Post-market clinical monitoring shall have one of two outcomes, (1) PMCFS required or (2) no PMCFS required.
The need for a PMCFS shall be based on a combination of several factors detailed in this section.
5.2.1.The Product Regulatory Affairs Representative in consultation with the Research
Manager or designee and Design Engineering and/or Engineering Representative
shall determine whether an equivalent device exists. Equivalence shall be
demonstrated in all the essential characteristics precisely defined below.
Equivalence means:
?Clinical
?Used for the same clinical condition or purpose;
?Used at the same site in the body;
?Used in similar population (including age, anatomy, physiology);
?Have similar relevant critical performance according to expected clinical effect for specific intended use
?Technical
?Used under similar conditions of use;
?Have similar specifications and properties;
?Be of similar design;
?Use similar deployment methods
?Have similar principles of operation
?Biological
?Same or similar use of materials in contact with human tissues or body fluids
5.2.2.Products for which the medium/long term clinical performance and safety is already
known from previous use of the device, or from fully transferable experience with
equivalent devices shall not require a PMCFS.
NOTE:If the device quoted as the “equivalent” requires a PMCFS, then the new
product shall be subject to the same requirement.
5.2.3.The need for a PMCFS shall be determined based on the identification of residual
risks that may impact the risk/benefit ratio. A study should always be considered
for devices where the identification of possible emerging risks and the evaluation of long term safety and performance are essential. The Product Regulatory Affairs
Representative in consultation with the Research Manager or designee and Design
Engineering and/or Engineering Representative shall identify such emerging risk, the following criteria should be taken into account:
?innovation, e.g., where the design of the device, the materials, the principles of operation, the technology or the medical indications are novel;
?high risk anatomical locations (i.e., heart, central nervous system, etc.);
?severity of disease/treatment challenges;
?sensitivity of target population (i.e., infants, children, pregnant women, etc.);
?identification of an acceptable risk during the pre-CE clinical evaluation, which should be monitored in a longer term and/or through a larger
population;
?well known risks identified from the literature or similar marketed devices;
?discrepancy between the pre-market follow-up time scales and the expected life of the product;
5.2.4. A properly conducted risk analysis is essential in determining what clinical evidence
may be need ed for a particular device. Any risks identified as an “unacceptable”
risk at the conclusion of the development process shall require a PMCFS. A study
should also be considered for risks identified as “acceptable” or “risk mitigation
required” if the de vice meets any of the other characteristics identified in 5.2.1 and
5.2.2. The risk assessment shall be performed according to the Risk Management
Procedure. The Product Regulatory Affairs Representative shall review the risk
assessment.
5.2.5.The Product Regulatory Affairs Representative shall complete the Post Market
Clinical Follow-Up Study Determination Form (Appendix A) once the decision
regarding the need for a study has been determined.
NOTE:This form may also be used as a guide in making the determination about the need to perform a PMCFS.
5.2.
6.The Product Regulatory Affairs Representative shall complete the Post-Market
Clinical Follow-Up Plan (Appendix B) that details the plan for post-market clinical
follow-up.
5.2.7.The Research Manager or designee and Medical Affairs Representative shall review
the Post-Market Clinical Follow-Up Justification Form and The Post-Market Clinical
Follow-Up Plan to confirm the decisions regarding post-market clinical monitoring.
5.3.No Post Market Clinical Follow-Up Study Required
5.3.1.If it was determined that no PMCFS is required (based on section 5.2), post-market
clinical monitoring is still required for the medical device.
5.3.2.Justification regarding the decision not to perform a PMCFS must be clearly
documented and maintained in the design history/technical file (see 5.2.5).
5.3.3.Post-Market Clinical Monitoring Requirements (minimum)
5.3.3.1.At a minimum, the following post-market clinical monitoring activities shall be
completed according to TDI Foot/Ankle Array 8ch established procedures/work
instructions. These elements will be inputs into the Post-Market Literature
Evaluation and Market Analysis Report.
?Review of product complaints according to Complaint Handling Procedure
?Review of post market adverse events according to Post Market Event Reporting Procedure
?Literature review according to TDI Foot/Ankle Array 8ch Evaluation of Clinical Data to Support CE Marking Work Instruction .
5.3.3.2.Review of product complaints, post market adverse events and the literature review
shall be completed at the intervals specified in Table 5-1. The timing outlined
provides the minimum requirements. The Product Regulatory Affairs
Representative and/or the Research Manager or designee can determine that
clinical data shall be reviewed more often.
Table 5-1: Timing for Review of Clinical Data based on Medical Device Class
5.3.3.3.At the interval outlined in Table 5-1, the Research Manager or designee shall
complete a literature review and analysis of post-market experiences (i.e.
complaints and adverse events) and re-evaluate if a PMCFS needs to be conducted
based on this data. The Post Market Literature Evaluation and Market Analysis
Conclusion form (Appendix D) shall be completed and maintained as part of the
device’s design history/technical file. The Product Regulatory Affairs
Representative and Medical Affairs Representative shall review and approve this
document.
NOTE:The literature review shall be executed according to the Evaluation of Clinical Data to Support CE Marking Work Instruction, section 5.5. However, the following forms/templates shall be used in place of those specified in this work instruction:
a.Instead of using The Literature Evaluation Plan template referenced, use the Post
Market Literature Evaluation and Market Experience Plan form (Appendix C)
b.Instead of using The Literature Evaluation Report and Conclusion template, use the
Post-Market Literature Evaluation and Market Analysis Report and Conclusion form
(Appendix D)
5.4.Post Market Clinical Follow-Up Study Required
5.4.1.If it was determined that a PMCFS is required, in addition to the requirements listed
under 5.3.3, studies such as extended follow-up of patients enrolled in the
pre-market trials, prospective study of a representative subset of patients after the
device is placed on the market, or an open registry may be performed.
5.4.2.The PMCFS shall b e carried out in accordance with TDI Foot/Ankle Array 8ch’s
Research Involving Human Subjects Procedure
5.4.3.The Research Manager or designee in consultation with the Regulatory Affairs
Representative and the Design Engineering and/or Engineering Representative will
determine the type of PMCFS that will be implemented.
5.4.4.The study should take into account the following:
?Results of the clinical investigation including adverse events identified
?Average life expectancy of the device
?The claims made by the manufacturer for the device
?Performances for which equivalence is claimed
?New information becoming available
5.4.4.1.At the interval outlined in Table 5-1, the Research Manager or designee shall
complete a literature review and analysis of post-market experiences (i.e.
complaints and adverse events) and review the ongoing results/data of the PMCFS.
The Post Market Literature Evaluation and Market Analysis Conclusion form
(Appendix D) shall be maintained as part of the device’s design history/technical file.
The Product Regulatory Affairs Representative and Medical Affairs Representative
shall review and approve this document.
NOTE:The literature review shall be executed according to the Evaluation of Clinical Data to Support CE Marking Work Instruction, section 5.5. However, the following forms/templates shall be used in place of those specified in this work instruction:
a.Instead of using The Literature Evaluation Plan template referenced, use the Post
Market Literature Evaluation and Market Experience Plan form (Appendix C)
b.Instead of using The Literature Evaluation Report and Conclusion template, use the
Post-Market Literature Evaluation and Market Analysis Report and Conclusion form
(Appendix D)
5.5.Elements of a post-market clinical follow-up study
5.5.1.Post-market clinical follow-up studies are performed on a device within its intended
use/purpose(s) according to the instructions for use.
5.5.2. A PMCFS shall include the elements defined in the Writing Clinical Investigational
Plans and Protocols Work Instruction.
5.5.3.The objective(s) of a PMCFS should be stated clearly and should address the residual
risk(s) identified. It should be formulated to address one or more specific
questions relating to the clinical safety or performance of the device.
5.5.4.Post-market clinical follow-up studies should be designed to address the objective(s)
of the study. The design may vary based on the objective(s) and should be
scientifically sound to allow for valid conclusions to be drawn.
5.5.5.The study design can take several forms, for example:
?the extended follow-up of patients enrolled in pre-market investigations;
? a new clinical investigation;
? a review of data derived from a device registry;
? a review of relevant retrospective data from patients previously exposed to the device.
?the analysis plan including any interim reporting; and
?procedures for early study termination.
5.5.
6.The data and conclusions derived from the PMCFS are used to provide clinical
evidence to support the post-market surveillance program. This process may result in the need to reassess whether the device continues to comply with the Essential
Principles. Such assessments may result in corrective or preventive actions.
6.APPENDIX
6.1.Appendix A: Post-Market Clinical Follow-Up Study Determination
6.2.Appendix B: Post-Market Clinical Follow-Up Plan
Experience Analysis Plan
Analysis Report and Conclusion
项目进度跟踪管理系统毕业设计(本科论文)
大连交通大学信息工程学院 毕业设计(论文)任务书题目项目进度跟踪管理系统
大连交通大学信息工程学院毕业设计(论文)进度计划与考核表 指导教师签字:: 2012年3月30日
大连交通大学信息工程学院 毕业设计(论文)外文翻译 学生姓名陈彬专业班级软件工程08-2班指导教师杨迪王立娟职称高级工程师讲师所在单位信息科学系软件工程教研室 教研室主任刘瑞杰 完成日期 2012 年 4 月 13 日 THE TECHNIQUE DEVELOPMENT HISTORY OF JSP
The Java Server Pages( JSP) is a kind of according to web of the script plait distance technique, similar carries the script language of Java in the server of the Netscape company of server- side JavaScript( SSJS) and the Active Server Pages(ASP) of the Microsoft. JSP compares the SSJS and ASP to have better can expand sex, and it is no more exclusive than any factory or some one particular server of Web. Though the norm of JSP is to be draw up by the Sun company of, any factory can carry out the JSP on own system. The After Sun release the JS P( the Java Server Pages) formally, the this kind of new Web application development technique very quickly caused the people's concern. JSP provided a special development environment for the Web application that establishes the high dynamic state. According to the Sun parlance, the JSP can adapt to include the Apache WebServer, IIS4.0 on the market at inside of 85% server product. This chapter will introduce the related knowledge of JSP and Databases, and JavaBean related contents, is all certainly rougher introduction among them basic contents, say perhaps to is a Guide only, if the reader needs the more detailed information, pleasing the book of consult the homologous JSP. 1.1 GENER ALIZE The JSP(Java Server Pages) is from the company of Sun Microsystems initiate, the many companies the participate to the build up the together of the a kind the of dynamic the state web the page technique standard, the it have the it in the construction the of the dynamic state the web page the strong but the do not the especially of the function. JSP and the technique of ASP of the Microsoft is very alike. Both all provide the ability that mixes with a certain procedure code and is explain by the language engine to carry out the procedure code in the code of HTML. Underneath we are simple of carry on the introduction to it. JSP pages are translated into servlets. So, fundamentally, any task JSP pages can perform could also be accomplished by servlets. However, this underlying equivalence does not mean that servlets and JSP pages are equally appropriate in all scenarios. The issue is not the power of the technology, it is the convenience, productivity, and maintainability of one or the other. After all, anything you can do on a particular computer platform in the Java programming language you could also do in assembly language. But it still matters which you choose. JSP provides the following benefits over servlets alone: ? It is easier to write and maintain the HTML. Your static code is ordinary HTML: no extra backslashes, no double quotes, and no lurking Java syntax. ? You can use standard Web-site development tools. Even HTML tools that know nothing about JSP can be used because they simply ignore the JSP tags.
ISO13485:2016医疗器械上市后临床跟踪程序
1、目的 适当使用和执行上市后临床跟踪研究,来解决与剩余风险相关的问题,及时修改产品标签,说明书的内容及技术文件,确保产品的持续安全有效,满足欧盟MEDDEV 2.12/2 rev2(《GUIDELINES ON MEDICAL DEVICES:POST MARKET CLINICAL FOLLOW-UP STUDIES A GUIDE FOR MANUFACTURERS AND NOTIFIED BODIES》January 2012发布)的要求。作为产品质量体系的一部分,一个适当的市场监督程序,对上市产品识别使用中风险识别和风险研究的关键,特制定本程序。 2、范围 适用于本公司带有CE标识的医疗器械产品的销售后的临床数据跟踪。 3、职责 3.1 管理者代表:负责组织落实产品符合MDD 93/42/EEC 的要求。 3.2 工程部:负责按本程序及相关法规制定《上市后的临床跟踪计划》及相关文件,并根据汇 总上市后的临床数据,评价现有文件的适合性并修改相关的文件。 3.3 品管部:负责按本程序及相关法规对文件归档保存,负责产品相关的标签的控制。 3.3 业务部:负责审核《上市后的临床跟踪计划》,并在计划批准后按文件要求执行。 3.4 总经理:负责对《上市后的临床跟踪计划》的批准。 4、定义 4.1 临床调查:在一个或更多的人类受试者上进行的任何系统性的调查或研究,从而评估医疗设备的安全性和性能。 4.2 上市后临床跟踪研究:跟随着设备的CE标记而执行的研究,其意图是回答与设备依照批准的标签使用时的临床安全性和性能(如剩余风险)相关的特定问题。 4.3 PMCF计划:制造商建立的备用证明文件的、前瞻性的、有组织的方法和程序,其根据与特定设计档案一致的CE标记产品的使用或属于相同亚类或MDD93/42/EEC指令中定义的一般设备组的一组医疗设备的使用来收集临床数据。它的目标是在医疗设备的预期生命期中巩固临床表现和安全性和经鉴定的风险的可接受性,以及在事实证据的基础上探测产生的风险。 4.4 剩余风险:风险控制措施后遗留的风险已被接受。
杭州MES系统产品追溯跟踪管理功能
中国制造业正面临技术升级的关键时刻,无论是机器换人、两化融合、智能制造、还是未来将实现“工业4.0”和“中国制造2025”,目前市场项目实施的关键是:MES系统+车间设备联网。“车间设备联网”是实现工厂“两化融合“的关键,可保证自动化层实时在线数据进入MES/ERP管理层。MES是实现车间级数字化管理的功能软件,MES与AI及互联网、云计算技术的融合是未来工厂智能制造的技术基础。 杭州,在这座城市中,总是充斥着大量的MES系统}相关信息,但是人们一时难以辨别其真假。 不要被小编的慷慨陈词所打动了,和你们说好做彼此的天使,所以今天不选择套路你们。好了,言归正传,来看看小编送上的MES系统福利是否能打动你吧~ 可追溯性是MES系统的一个重要特性,可追溯数据模型不仅可以完整记录生产过程数据,还可以扩展到质量追溯、采购追溯等方面,对企业制造过程控制和制造过程改进具有重要意义。在生产车间仓工人将产品放错是很正常的事情。MES追溯管理系统主要是帮助企业进行产品生产基础数据整理、物料防错管理还有产品整个生产销售流程的追溯管理。预防人为因素造成工艺漏装。 MES防错追溯管理系统主要是使用统一的信息管理方法,在装配线上通过安装一维/二维 条码、RFID等信息载体。华磊迅拓的MES系统软件通过扫描枪的实时扫描和对比,通过一体机或触摸屏识别装配件是否符合要求,以及将装配过程中的实时数据发送到系统记录服务器。 MES防错追溯管理系统主要功能
1.生产计划导入 (1)计划管理员制订每条生产线的上线计划,把计划录入防错系统计划导入模板文件; (2)进入防错系统上线计划导入页面,选择上传计划后,系统把计划数据导入用户界面,同时后台并检查计划内容是否正确; (3)确认计划数据无误后确认,防错系统把计划导入系统数据库; (4)可以实现上线批次的顺序、计划数量操作,对于删除和更新的计划,系统记录计划变更日志,在计划追踪查询中给予标注; (5)计划调整时允许多行选择计划的删除功能; (6)提供一个操作软件,安装于办公室PC机,联网运行,由有权限的操作员执行。 2.MES商品质量防错 MES防错系统是检查在装配过程中一些关键步骤或关键操作是否成功完成,包括制造过程中重要零件符合性检查和精确追溯信息的管理,零件追溯实际对生产物流控制提出流程要求和数据要求,通过对追溯物料的状态跟踪来控制质量遏制范围。通过扫描枪读取零件条形码用以检查零件是否和显示的部件相符,以防止错装。指导车间生产,可进行生产流程完工反馈。 系统监控关键件装配,预防人为因素造成工艺漏装。可规范产品作业流程,并严格要求按产品流程作业。解决了门板装配过程中缺少装配作业的问题,做到实时监控、及时反应,立刻解决。这个小编之前还跟大家提过包装称重防错系统,也是一样的道理。 3.MES防差错操作流程 (1)同一台终端设备根据订单类型自动显示不同的装配界面指导装配作业; (2)按照工艺流程要求严格把关装配过程,装配人员只需按照系统提示进行装配,排除人为因素造成的潜在风险,促使装配过程标准化、透明化; (3)装配数据必须符合装配零件校验规则,系统对每个扫描的零件条码进行验证,若不符合零件校验规则则自动报警提醒并禁止下一步工艺装配; (4)MES系统通过解析关键件、重保件的条码与工序进行匹配,从而实现质量防遗漏、质量防差错、质量数据正、反追溯; (5)完成整个工序过程自动放行并显示下一个订单配置、装配信息。 4.零件校对规则 对装配零件与数据进行有效性较验规则进行设置。可根据供应商提供的子零件不同的条码规则(如扶手、装饰条、前门总成、后门总成)在MES系统中配置,配置数据将作为MES对装配过程中的零件进行防差错较验。
2020版药物临床试验质量管理规范试题
2020版药物临床试验质量管理规范试题 1. 以下哪个法律法规不是《药物临床试验质量管理规范》制定的根据: A.《中华人民共和国药品管理法》 B.《中华人民共和国疫苗管理法》 C.《中华人民共和国药品管理法实施条例》 D.《药品生产监督管理办法》 2. 研究者在临床试验过程中应当遵守试验方案,凡涉及医学判断或临床决策应当由以下哪个角色做出? A.临床试验协调员 B.临床医生 C.伦理委员会 D.监察员 3. 以下哪项不是临床试验的质量管理体系的重点? A.受试者保护 B.试验结果可靠 C.试验药物潜在收益 D.遵守相关法律法规 4. 独立的数据监查委员会(数据和安全监查委员会,监查委员会,数据监查委员会)由谁设立? A.申办者 B.研究者 C.试验中心 D.药政部门 5. 告知一项试验的各个方面情况后,受试者自愿认其同意参见该项临床试验的过程是: A.知情同意 B.知情同意书 C.试验方案
D.研究者手册 6. 通过签订合同授权,执行申办者或者研究者在临床试验中的某些职责和任务的单位是什么? A.伦理委员会 B.监查员 C.协调研究者 D.合同研究组织 7. 受试者被告知可影响其做出参加临床试验决定的各方面情况后,确认同意自愿参加临床试验的过程。该过程应当以书面的、签署姓名和日期的文件是: A.研究者手册 B.试验方案 C.知情同意书 D.标准操作规程 8. 对临床试验相关活动和文件进行系统的、独立的检查,以评估确定临床试验相关活动的实施、试验数据的记录、分析和报告是否符合试验方案、标准操作规程和相关法律法规的要求的行为是: A.监查 B.稽查 C.检查 D.直接查阅 9. 受试者接受试验用药品后出现死亡、危及生命、永久或者严重的残疾或者功能丧失、受试者需要住院治疗或者延长住院时间,以及先天性异常或者出生缺陷等不良医学事件指的是: A.不良事件 B.药物不良反应 C.严重不良事件 D.危险信号 10. 以下对于伦理委员会的组成和运行描述不正确的是: A.伦理委员会的委员组成、备案管理应当符合卫生健康主管部门的要求。 B.伦理委员会的委员均应当接受伦理审查的培训,能够审查临床试验相关的伦
调查方法与技巧 关于跟踪和反跟踪
调查方法与技巧关于跟踪和反跟踪 2007-07-28 23:32:50 作者:陈利发表评论 (作者简介: 陈利,南昌市人民警察学校高级讲师、中国公安优秀教官、江西法制心理专业委员会委员、南昌市公安局特邀研究员,浙江万马公众事务调查中心总技术指导.) 在人类社会中,一切事物都有其产生、存在和发展的客观条件和规律,都有它本身固有的特性,以区别于其他事物。为此,我们研究调查业时,就必须探讨调查活动的方法和技巧。 一、徒步跟踪调查 调查准备工作 当调查人员使用常规的调查手段,得不到更多的资料时,就要考虑实施人员监视来获取材料。实施人员监视时,调查人员将直接观察想要了解的情况,包括设法取得有关特定的证据;如有可能,要设法对调查对象正在进行的行为进行直接观察,使用任何种类的人员监视行动的真正目的,是取得有关材料或证据。监视行动成功与否,主要是根据在实施监视行动之前,制定的预定行动计划的详尽和慎重的程度。无论是刑事案件还是民事案件,无论当时的情况是需要运动监视、步行监视或乘车监视、还是定点监视,预定计划的重要性是无可非议的。 1、实施人员监视时,调查人员必须考虑到,在将要实施的实际监视工作中,调查工作本身和各种环境的特定要求。为了获取一般需要的材料,例如对象的习惯、社会交往以及生活方式,是采取一般性尾随监视呢?还是因为案件性质的敏感性和需要时刻密切观察对象的活动,采用紧紧咬住的监视?如果要想保证监视行动有个令人满意的结果,调查人员要考虑到这些客观因素。制定出的方案与实际情况不符合,要么导致监视行动失败,要么是在时间与经费上作出不必要的支出。制定出的监视行动方案的范围包括:根据案情所需要的调查人员数量;整个监视过程中需要使用的技术装备;以及可能会出现的疵漏问题;实施运动监视时随身携带数量充足的现金,这包括小面额钞票和零钱,这样才能确保对象无论选用什么手段运动到任何地方,调查人员都能方便地尾随其后。 2、初步调查的重要性,再怎么强调也不过分。尤其是在处理极为敏感的案子时,用两名调查人员分别进行初步调查的事屡见不鲜。然后公司对这两个调查方案进行检查核实,在制定计划时作为参考的依据。基本上可以这么说:调查人员首先应该明确地确定一下,自己需要的是什么材料和情报,然后再设法开展一段时间的监视行动。只有这样,才有希望得到所需的情报。当调查人员对自己是否能搞到所需要的情报感到到信心不足、抱着一种怀疑态度时,使用这种方法的话,就能使调查人员在观察对象期间,不会浪费时间。在许多要求取得对象背景材料的案例中,已证明这种办法是行之有效的。为了获得对象的背景材料,调查人员要耗费大量的时间。 3、准备工作阶段,调查人员应设法确定出实施监视的最佳期时间。在任何行动中,对某人监视的时间越长,引起对象警觉的可能性就越大。因此,在制定有效的监视行动方案时,要考虑到两个最佳值得引起注意的问题,尽量减少被对象侦察出的机会和把时间与经费降低最低限度。调查人员在进行初步调查时,要挑选几套式样与监视地区情况相符的衣服。如果实施运动监视,对实施监视行动的整个地区都进行一次调查,而且对对象可能将要通过的地区也进行调查,实施完初步调查之后,调查人员对于需要使用什么器材,心中就完全有数了,并能决定使用哪一类器材,如望远装置、夜视仪或照相机。 4、监视人员在结束监视行动时,要写一份书面报告总结整个监视过程,因此在制定计划时也要有相应的思想准备。初步调查任何监视行动,无论是运动监视还是定点监视,在制定方案期间,调查人员对将要监视对象的地区、或第一次看到对象的地点,最好进行-下初步调查,例如:如果是在对象清早离开家时实施运动监视,调查人员对对象的邻居实施事前调查,
任务信息管理系统需求分析说明书案例参考样本
技术文件 文件名称: 任务管理系统需求说明书项目名称: 任务管理系统 共页 (包括封面) 作者:
1 引言 1.1 编写目的 本文详细描述任务管理系统的需求, 表述的需求信息要求明确、无二义性。开发方与软件使用者充分沟通需求, 最终形成此文档。此文档是后续软件开发的依据。 1.2 背景 任务管理系统是一个XX与XX电气新技术有限公司产学研合作项目, 项目由XX机电新技术有限公司提出, 由XX承担开发任务。 1.3 定义和缩略语 本文使用了错误!未找到引用源。所显示的面向用户的术语、定义, 包括通用词语在本文档中的专用解释。 表 1.1 术语/定义 错误!未找到引用源。所列为本文用到的缩略语。 表 1.2 缩略语
1.4 参考资料 本文使用了错误!未找到引用源。所列为本文用到的参考资料。 表 1.3 参考资料 1.5 用户 任务信息管理系统的当前用户为XX公司电气事业部, 电气事业部使用成功后可能会在XX公司推广。 2 任务概述 2.1目标 XX公司电气事业部当前的任务主要有2类: 常规工作任务和临时性工作任务。 针对临时任务布置信息很多时候是处于一种开放状态, 缺少任务信息的修正、回馈、和统计分析。而日常职责规定的常规工作, 虽然能够经过标准化的文件固化下来并形成《常规工作计划表》作为一种制度来执行, 也需要主管在百忙之中花很多时间去检查完成情况。 TIMS系统要求工作管理信息能够规范录入, 任务信息流向能够选择, 任务信息依据轻重排序, 能够设定信息提醒, 任务完成
情况能够评估、任务完成情况依据选择项进行统计输出、工作量进行评估。 2.2 系统的特点 TIMS项目的需求主要由XX公司电气事业部提出, 因此本文档是与XX公司电气事业部交互后形成的需求定义, 系统的功能和使用特点优先满足XX公司电气事业部的需求, 若系统后续由于在XX 公司全面推广而引入的新需求, 则不在本文档考虑范围之内。 2.3 假定和约束 本文档经双方确认后, 开发方依据本文档进行下阶段工作。若中途需求发生变更则XX公司需及时告知开发方, 若因XX公司原因引入的需求变更造成开发方工作量的大幅增加, 具体解决方案双方另行协商。若需求变更引入的工作量不大, 开发方应尽量配合。 4. 需求规定 4.1 组织架构 XX公司电气事业部的组织架构如图4-1。
澳大学倒闭事件持续追踪解决方案已公布
澳大学倒闭事件持续追踪解决方案已公布 一位女生担心前途,当众落泪 事件追踪澳大学倒闭影响近千中国学生 澳大利亚4所私立大学5日因破产闭校,致使约2700名外国留学生失学。这4所大学位于悉尼和墨尔本,为全球校园管理集团所有,统一名称为莫瑞迪安国际酒店管理学院。该集团5日宣布,4所大学因破产进入自愿托管阶段。学校多为亚洲留学生,包括近千名中国学生。中国驻悉尼和墨尔本领馆已要求有关部门确保中国留学生的利益。澳政府官员说,准备将失学学生转送到其他学校以帮助他们完成学业。 突然闭校 南京约有10余名留学生遭遇失学 学生们事前未得到任何关闭学校消息 政府表态 尽快帮助学生转学重返课堂 9日将公布事件具体解决方案 倒闭原因 因合伙人撤资引发学院破产 5月份来已有10家私教机构倒闭 发展 中国领事馆提出交涉
中国驻悉尼总领馆教育处白刚参赞已向澳有关教育主管部门和移民局等提出交涉,敦促澳方妥善处理。目前使馆官员已与部分留学生见面,就如何表达自己诉求进行了指导,并告知相关学生向澳方投诉的渠道和联系方式。澳方表示将积极、妥善处理好此事,私立教育培训委员会已经通过电子邮件的形式通知学生,他们将在11月10日和11月11日了解学生们的诉求,尽快安排学生转学、退学费等事宜。》》详细 解决方案 将按照学费保险计划的程序来处理 白刚:今天早晨我们在9点30分已经向澳大利亚私立教育培训委员会进行了交涉,他们提到将按照学费保险计划的程序来处理这件事情。澳大利亚私立教育培训委员会已经通过电子邮件的形式通知学生,将在11月10日和11月11日召开学生会议,他们准备了解学生们的诉求,尽快处理这件事情。 倒闭学校或由政府接管———私立变为公立 澳大利亚教育部长朱莉娅·吉拉德6日发表声明,政府将帮助失学学生渡过困境。 据艾迪国际南京分公司邢刚经理了解到的最新消息显示,针对这四所倒闭的私立院校,学生的安置工作将由澳大利亚教育、就业及劳动关系部负责全程监督,具体方案将在11月9日出台,初步估计学校将会由政府接管,私立变为公立,或是现读学生转入其他公立院校。邢刚经理认为,鉴于澳洲完善的教育管理体系,私校倒闭给学生带来的影响不会很大,在这次倒闭事件中受到影响的学生,其文凭以及后续的教育也会获得澳洲政府的妥善安排。 声音 学生:如果澳政府不解决我们将会采取行动 记者:现在算来失学已经有三天时间了,你现在在做什么,和你一样有同样遭遇的同学他们的情况怎么样了?
MEDDEV 2.12-2 医疗器械上市后临床跟踪指南
EUROPEAN COMMISSION DG ENTERPRISE Directorate G Unit 4 - Pressure Equipment, Medical Devices, Metrology MEDICAL DEVICES: Guidance document MEDDEV 2.12-2 May 2004 GUIDELINES ON POST MARKET CLINICAL FOLLOW-UP 上市后临床跟踪指南 The present Guidelines are part of a set of Guidelines relating to questions of application of EC-Directives on medical devices. They are legally not binding. The Guidelines have been carefully drafted through a process of intensive consultation of the various interested parties (competent authorities, Commission services, industries, other interested parties) during which intermediate drafts were circulated and comments were taken up in the document. Therefore, this document reflects positions taken by representatives of interested parties in the medical devices sector. 本准则是一个有关的欧共体指令对医疗设备的应用问题指引的一部分。他们在法律上没有约束力。该指引已审慎草拟通过各有关方面(主管机关,委员会的服务,工业,其他有关各方)在此期间,中间草案分发和评论的文件采取了密集的磋商进程。因此,这份文件反映了有关各方的代表在该领域采取的医疗设备的位置。 Foreword : Rationale and Goals of PMCF This document is intended to be a guide for manufacturers and notified bodies on how to carry out PMCF in order to fulfill post market surveillance obligation according to point 3. 1 of annex II, point 3. of annex IV, point 3 of annex V, point 3.1 of annex VI or point 4 of annex VII of medical device directive (add ref. AIMDD) While clinical evidence is an essential element of the premarket conformity assessment process, it is important to recognize the limitations inherent to these premarket clinical investigations. The extent of the data that can be gathered in the premarket phase does not enable the manufacturer to detect infrequent complications or problems only apparent after widespread use, or /long term performance issues. As part of the manufacturer’s quality system, a program of appropriate post market surveillance is key to identifying and investigating risks associated with the use of medical devices placed on the market. 前言:PMCF的基本原理和目标 本文件的目的是为制造商的指引,就如何开展PMCF为了履行市场监督义务后按3点通知机构。1附件二点三。附件四,附件三点五,六31点附件或附件4点的医疗设备指令(七加号。AIMDD)
(完整版)任务管理系统需求分析
项目名称:某企业任务管理系统
1. 项目背景及其需求 1.1 项目背景 大唐软件技术有限责任公司(CATTSOFT)(以下简称“大唐软件”)是大唐电信科技股份有限公司的全资子公司。大唐软件以提供适合各通信网络和通信业务运营商需要的管理软件、支撑软件、增值业务软件系统为业务基础,为各类通信系统运营商或信息系统用户提供业务管理、网络管理、决策支持、系统集成和专业咨询的完整解决方案和服务。 现承接大唐软件某业务部门的“业务管理系统”中“任务管理系统”子系统的设计和开发。 1.2 系统需求 1.2.1 术语解释 1.2.1.1 系统管理员 是该系统的一种用户,其权限是添加其他用户并分配其角色(包括主管和员工)。1.2.1.2 主管 是该系统的一种用户,一个主管下属有一些员工。主管的主要权限是创建任务描述,并将该任务分配给其下属的员工。主管还可以跟踪任务的实施情况。 1.2.1.3 员工 该系统的一种用户,其主要权限是将上级主管分配的任务分解为具体的实施计划。再必要的时候可以调整计划的内容。 1.2.1.4 任务 任务是由主管创建并分配给员工的一项工作。一个任务有“待实施”、“实施中”和“已完成”三种状态。当主管建立一个新任务时,该任务的状态为“待实施”;当承担该任务的员工为该任务制定了计划后,可以将该任务的状态改为“实施中”;主管通过任务跟踪,当认为任务已经完成时,可以将该任务的状态改为“已完成” 1.2.1.5 计划 是由员工创建,表示一个任务的具体实施过程。一个任务可以对应多个计划,计划有两种状态“未反馈”和“已反馈”。当计划刚刚建立时,其状态为“未反馈”,当计划已经完成时,员工可以填写反馈信息并将其状态改未“已反馈”。
Win Server2003常见问题及解决然方案
Win Server2003常见问题及解决然方案 2005-12-10 14:59:25 作者:来源:不详浏览次数:119 评论 随着windows server 2003的上市在即,很多人可以用上的泄漏的版本,相对于工作站系统,服务器在由于做了更多的内核优化,所以在稳定性和安全性方面有很大的提高。但是,很多人并不是需要Server的全部功能的,而且Server系统关闭了某些工作站系统所需要的服务,下面,我们将对如何优化windows server 2003并转换成一个工作站系统做出一些介绍。1.禁用配置服务器向导 由于不需要服务器设置功能,首先我们先禁止“配置你的服务器”(Manage Your Server)向导的出现,你可以在控制面板(Control Panel) -> 管理员工具(Administrative Tools )-> 管理你的服务器(Manage Your Server)运行它,然后在窗口的左下角复选“登录时不要显示该页”(Don''t display this page at logon)。 2.建立一个新的用户账号 windows server 2003不支持类似于Windows XP的登录欢迎屏幕。你可以在首次进入系统后建立一个有你个性的新用户账号。 打开“开始”(Start) -> “运行”(Run) -> 键入“lusrmgr.msc ”,你将看到本地用户和组(Local Users and Groups ), 右键点击左边窗口的“用户”(Users),选择“新用户”(New User).在弹出的对话框中输入账号信息,然后点击“建立”(Great)。这样你的账号就可用了,当然,你可以选择把你的账号添加到管理员组,右键点击你新建的用户。然后选择“属性”(Properties) -> 点击“隶属于” on Member of tab -> “添加”Add.. -> “高级”Advanced -> “现在查找”Find Now 。 在查找结果对话框中双击“管理员”(Administrators),在点击两次“确定”(Ok)后关闭“本地用户和组”(Local Users and Groups window ),现在你将可以注销Administrator 用户用你自己的账号登录系统。 3.禁用Internet Explorer Enhanced Security 作为新windows组件出现的IE安全插件--Internet Explorer Enhanced Security默认把你IE安全设置为最高。这样你将在访问站点弹出询问框并对你浏览网页及文件下载做出阻止的行为。我们其实不一定需要这个组件。 我们首先禁止询问框的出现,在弹出的对话框中复选”以后不要显示这个信息“ (In the future, do not show this message) 然后,我们可以在IE工具选项中自定义设置IE的安全级别。在”安全“(Security)选项卡上拉动滚动条把Internet区域安全设置为”中“(Medium),这个级别将适合大多数人,
临床跟踪总结报告-模板
产品名称 临床跟踪总结报告 公司名称 日期
目录 一、临床跟踪的背景 (1) 1.1 临床跟踪产品背景信息 (1) 1.2 产品的作用机理和原理 (1) 1.3 产品技术特点 (1) 1.4 产品性能特点 (1) 1.5 产品使用特点 (1) 二、临床跟踪的目的 (1) 三、临床跟踪计划 (1) 3.1 适用范围及研究问题 (1) 3.2 临床跟踪研究方法 (1) 3.2.1 纳入标准 (1) 3.2.2 排除标准 (1) 3.2.3 实施临床跟踪的机构 (2) 四、结论 (3) 附件一XXX医院临床数据汇总 .................................................................... 错误!未定义书签。附件二XXX医院临床跟踪数据汇总 ............................................................. 错误!未定义书签。附件三XXX医院临床跟踪数据汇总 ............................................................ 错误!未定义书签。
一、临床跟踪的背景 1.1 临床跟踪产品背景信息 产品用于…… 行业现状说明 1.2 产品的作用机理和原理 1.3 产品技术特点 1.4 产品性能特点 1.5 产品使用特点 二、临床跟踪的目的 通过对我公司生产的XXXX上市后的临床跟踪,建立该产品临床性能的长期安全性和有效性,确定风险的可控性,并且能评估实际临床应用过程中可能出现的风险。 三、临床跟踪计划 对已上市的产品实施临床跟踪计划,通过对已销售到市场上的XXXX进行临床跟踪,证明该产品的安全性和有效性。 3.1 适用范围及研究问题 适用范围: 通过研究以解决如下问题: ●在临床应用过程中是否存在风险管理中所提及的风险; ●XXXX长期在市场上使用过程中的安全性及有效性; ●在临床应用过程中是否存在新的风险。 3.2 临床跟踪研究方法 3.2.1 纳入标准 ●5岁以上(含),75岁以下(含),无性别要求。 ● 3.2.2 排除标准 如有下列情况的,患者不应被进行临床跟踪: ●患者不符合上述入选准则的;
文件与记录管理程序
文件与记录管理程序 1、目的 通过对管理体系文件的严格控制管理,确保其文件使用的有效性、保管和更改的规定。并对质量记录其完整性、准确性、清晰、保管等予以控制。以证明产品质量满足规定要求、管理体系有效运行,并为实现可追溯性、证实作用以及采取纠正和预防措施提供依据。 2、适用范围 适用于本公司所有管理体系文件及相关资料记录的管理与控制。 3、职责与权限 3.1 ISO文控中心负责公司管理体系文件的收集、分类编号、整理、存档保管、发放及与管理和 控制有关的其他工作。 3.2 各部门负责本部门在用文件的管理和控制。 4、程序内容 4.1 管理体系文件的分类、编制、审核、审批按下列规定执行: 4.2 管理体系文件的编号、归口、分类、 4.2.1 公司编制的各级管理体系文件均由文控中心归口、分类并统一编号,其文件编号规定 4.2.2 文件受控分类 管理体系文件分为受控原版、受控副本、非受控副本: 1) 受控原版是不须加盖受控文件印章,由文控中心保存,仅作为复制用途。 2)受控副本为受控原版的复印件,其上加盖红色“受控本”印章发放,作为公司内使用的合法文件,公司内不允许使用“受控本”变黑的非法复印文件,以确保文件更 新时能够从发放或使用场所撤出失效或作废的文件。 3)非受控副本为受控原版的复印件,其上加盖红色本公司名印章作为提供给客户或公司以外的人员使用的合法文件。公司内部不允许使用加盖红色本公司印章名的体系 受控文件。 4.2.3 文件版次规定 为确保公司管理体系文件的有效性能够得到控制,对其采用版次来控制最新有效版次。版次采用“A”+第几次版本,文件经过一次修改后进行换版,版本号增高一位(如: A0首次发行版本、A1第一次修改、A...n第n次修改 4.3 文件的发放与记录 4.3.1 文控中心根据质量手册、程序文件、质量记录制定并及时登录《受控文件总表》;根 据质量记录表格制定并及时登录《质量记录总表》,确保各部门使用最新有效的文件。
用泛微OA系统进行任务管理,实现高效的任务跟踪与反馈
移动办公专家https://www.360docs.net/doc/f29354697.html, 用泛微OA系统进行任务管理,实现高效的任务跟踪与反馈 身为领导,总是免不了与任务部署打交道,然而因缺乏高效的任务反馈与跟踪体制,有时候往往出现啼笑皆非的状况: 布置下去的任务,就如丢入大海的石头,一不留神儿就渺无音讯。如何才能摆脱这种尴尬处境实现高效的任务跟踪与反馈呢?泛微e-office OA系统通过高效的任务管理,实现全方位任务分配、跟踪、反馈,让领导管理任务更轻松,员工汇报进度更及时。 随时随地分配任务
移动办公专家https://www.360docs.net/doc/f29354697.html, 移动OA实现随时随地新建、分派任务,解决差旅途中任务分派问题。新建任务时,可指定具体负责人,并明确相关参与人,以便分工合作进行团队协作。除此之外,还可关注下属自建的任务,并随时查看进度情况。 明确任务时间节点
移动办公专家https://www.360docs.net/doc/f29354697.html, 在新建任务时,通过设置到期日,进行时间节点的控制。同时,为了防止紧急的、重要的事项被淹没可设置紧急程度,确保重要事项优先处理。 细化任务循序推进 为确保任务的有序推进,可将其细化,建立与之相关的子任务。一个任务可对应多个子任务,并明确每个子任务的负责人。通过这样的设置,专人做专事,整体任务得以高效推进! 及时掌握任务进度
移动办公专家https://www.360docs.net/doc/f29354697.html, 分派下去的任务迟迟没有反馈不知道进展如何了,登陆手机任务管理及时查看!通过查看“任务分析”,以图表的形式清晰展现与任务相关的进度情况、完成情况。 除此之外,还可进入“下属任务”查看某项任务的具体进度条,结合时间节点与进度条对下属进行催办、督促等。每一个任务下的子任务情况清晰展示,进一步确保对任务整体情况的掌控。 评估任务执行情况 任务结束,上级可对下级的任务执行情况、完成质量等进行评分。通过评估此次任务中下属的表现,对以后任务的分配以及月末的考核起到参考作用,建立良好的人才机制。 通过泛微OA系统进行任务管理,团队的目标经过层层细分,最终形成具体到团队每个成员每天的具体任务。有效的任务管理是实现企业目标的重要手段,也是细化企业管控的重要环节。
事件管理流程
事件管理流程
目录 事件管理流程 (1) 1目的 (3) 2适用范围 (3) 3术语与定义 (3) 4流程客户 (5) 5输入与输出 (5) 6主要角色及职责 (5) 7普通事件管理工作程序 (6) 8关键步骤说明 (6) 9事件跟踪 (8) 10重大事件管理工作程序 (9) 11关键步骤说明 (9) 12流程质量控制 (11) 13与其他流程的接口 (11) 14关键业绩指标 (11) 15支持性文件 (11)
1目的 事件管理流程旨在介绍和描述事件管理流程,并将作为XXX公司日常IT维护所涉及的事件管理流程的参考。本文介绍了如何在事故发生、诊断、到关闭的整个生命周期中实施事件管理,并定义支持运作事件管理流程相关的人员职责。 事件管理流程提供了事件管理的步骤和规范,以满足事故解决的准确性、及时性和有效性。 事件管理的目标是尽快恢复协商一致的服务或者响应服务请求。 事件管理需要保留事件的有效记录以便能够权衡并改进处理流程,给其他的服务管理流程提供合适的信息,以及正确报告进展情况。 2适用范围 XXX 3术语与定义 事件 事件是在服务过程中发生的任何不符合标准操作且已经引起或可能引起服务中断和服务质量下降的事态,同时事件不仅包括软硬件故障,还包括服务的请求。 服务台 服务台是目标客户与技术服务工程师和专家进行联络的单一接合点。 高效的服务台不仅能节省大量的人力、物力、财力,在快速高效地为用户解
决问题的同时,更有有效地增加客户粘合度。 影响度 影响度指就所影响的用户或业务数量而言,事件偏离正常服务级别的程度。 紧急度 紧急度指解决故障时,对用户或业务来说可接受的耽搁时间。 优先级 主要基于紧急度和影响度来决定。而对于具有同样优先级的事件,可按解决他们需花费的精力的多少来安排顺序。例如,对某个影响不大且容易解决的故障,可先于一个影响较大且需要大量精力解决的故障。 升级 如果某一事件不能在规定的时间内由一线支持小组解决,那么更多有经验的人员和有更高权限的人员将不得不参与进来。这就是升级,它可能发生在事件解决过程的任何时间和任何支持级别。升级分为职能性升级和结构性升级。 重大事件 重大事件是指那些对用户团体带来非常严重影响的事件。而有些在时间上极度紧迫的需要解决的事件也应当作重大事件来处理。 事件状态 事件在处理过程中所处的各种状态,用来标注事件处理的程度。详细的状态定义详见《事件管理实施细则》
跟踪管理系统用户手册
消 防 产 品 跟 踪 管 理 系 统 生 产 企 业 客 户 端 简明用户手册 公安部消防产品合格评定中心 制作单位 北京优士东方数码科技有限公司 制作日期: 二零零七年十二月
前言 首次安装消防产品跟踪管理系统,基础信息下载完毕后,请您立即通过系统升级获得最新版本软件,以保证您所使用的软件版本同服务器保持一致。 阅读手册 欢迎您使用消防产品跟踪管理系统。 通过阅读本手册,您可以快速了解如何安装和使用消防产品跟踪管理系统,以及一些产品安装和使用过程中的注意事项。为帮助您更好地阅读,现将手册的内容结构、所用约定及特定术语的含义,简单介绍如下: 手册内容结构 手册共分四部分: 第一部分 前言 介绍手册使用相关问题和系统简介,本产品使用过程中的一些注意事项; 第二部分 产品安装 介绍产品的安装方法; 第三部分 操作说明 生产厂家使用本产品时的基本概念及主要操作流程; 手册中的术语 z准入标志 公安部消防产品合格评定中心颁发,采用专业印刷技术生产出的精密数字标识。每个准入标志具有唯一性。准入标志只能由通过消防产品准入生产评定审核的产品生产厂家粘贴在被批准的产品型号、并且必须由生产厂家进行准入标志信息启用注册机才能正式使用。目前分为准入标志I和准入标志II两种规格。 z专用读取设备 本系统采用UD笔作为准入标志的信息专用读取设备。UD笔具有与普通笔一样的纸面手写功能,而且具有在数码纸面上读取手写信息并进行存储和传输的功能。每套专用读取设备都具有唯一的设备号。只有经过评定中心发放认可的UD笔才能使用。 z数码表单 本系统使用的标签启用注册表、监督检查表是采用专用数码纸印制的,各单位根据需要购买经评定中心认可的产品。 系统简介 消防产品跟踪管理系统是以在数码纸笔基础上建立起来的标识技术为核心,结合其他防伪技术,根据消防产品在生产和监管过程中的使用特点,对消防产品进行全过程监管的综合管理系统。