生物序列联配中的算法全版.ppt
序列分析(二)

例:
((LYCES, SPIOL 84), (YEAST, (XENLA, (((RAT, MOUSE 96), HUMAN 83), CHICK 71) 66), DROVI 58))
相关树
多序列比对
目前使用最广泛的多重序列比对程序是 ClustalW
– ClustalW是一种渐进的比对方法,先将多个序 ClustalW是一种渐进的比对方法,先将多个序 列进行两两比对,基于这些比较,计算得到 一个距离矩阵,该矩阵反映了每对序列的关 系
多重序列比对投影
2、多重比对的动态规划算法
• 多重序列比对的最终目标是通过处理得到一个得分最
高(或代价最小)的序列对比排列,从而分析各序列 或代价最小)的序列对比排列, 之间的相似性和差异。 之间的相似性和差异
前趋节点的个数等于2 前趋节点的个数等于 k - 1
假设以k维数组 存放超晶格 则计算过程如下: 假设以 维数组A存放超晶格,则计算过程如下: 维数组 存放超晶格, a[ 0, 0, … ,0 ] = 0 a[ i ] = max {a[ i - b ] + SP-score(Column(s, i, b))}
另一种计算方式: 另一种计算方式:先处理每一个序列对 在处理序列对时,逐个计算字符对, 在处理序列对时,逐个计算字符对,最后加和 得分模型的计算公式如下: 则SP得分模型的计算公式如下: 得分模型的计算公式如下
SP − score(α ) = ∑α ij
i< j
α 是一个多重比对 αij是由α推演出来的序列 i 和s j的两两比对 是由α推演出来的序列s
∑ sim( si, sc )
时间复杂度为O 时间复杂度为O(k2n2 + kn2)
细菌全基因组测序 ppt课件
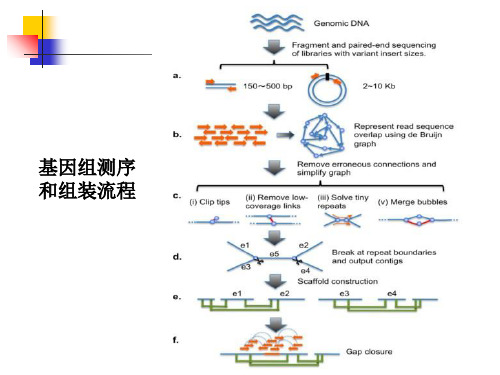
基因家族(gene family) 和基因簇(gene cluster)分析
基因组中来源相同,结构和功能相关的基因 聚集在一起形成基因家族。
基因家族的各个成员紧密成簇排列成大段的串联 重复单位,分布在某一条染色体的特殊区域
genefamily.xls
基因家族聚类结果
genefamily.stat
各基因家族统计信息
培养条件① 培养条件②
或活性较低
测定转录 组mRNA
细菌全基因组测序
比较 新 差异 基因
其他方面的应用研究
❖ 应用NMR、FTIR、UV, 14C标记的木质 素降解机理方面的研究; ❖农药残留物以及其他一些难降解有机物的 降解; ❖ 重金属有机物化合物的降解。
② 木质素降解过程中涉及到的细胞外酶主要有:木质素过氧化物酶
(LiP)和锰过氧化物酶(MnP),以及漆酶(Lac)。此外,一 些附属酶参与过氧化氢的产生,乙二醛氧化酶(glyoxal oxidase, 缩写作GLOX)和芳基醇氧化酶(aryl alcohol oxidase,缩写作 AAO)属于这类酶。
对4株菌的亲缘关系进行分析,确定菌株之间的相互关 系;
通过对4株菌进行进化分析,判定是否为古菌或新的菌 种。
细菌全基因组测序
基因分离
下一步的实验安排
对已注释出的基因进行验证
载体
酶切
酶切
连接
转化
筛选 表达
细菌全基因组测序
未注释出功能的基因鉴定,挖掘新基因
DNA 转录 RNA 翻译 Protein
细菌全基因组测序
“一个物种基因组计划的完成, 就意味着这一物种学科和产业 发展的新开端”
向仲怀院士
谢谢!!
细菌全基因组测序
生物信息学 第四章 序列比对与算法

T
C A G A T 1 1 1
1
1
1
1 1 1 1 1 1 1 1 1
1
1
1
1
A A C C G T C A G A T 1 0 A A C C G T C A G A T 4 3 4 0 1 1 1
C
T
G
C
T
A 1
A 1
A 1
C
G
T A
A 1
C
T
G
C
T
A 1
A 1
A 1
C
G
T
1 1 1 1 1
矩阵的元素表示由原来的氨基酸(上行,蓝色)替换为另一个氨
基酸(左列,绿色)的概率
例
Score with PAM 250 and gap penalty -10
计算如下比对
CKHVFCRVCI CKKCFCKCV
CKHVFCRVCI CKKCFC-KCV CKHVFCRVCI CKKCFCK-CV C-KHVFCRVCI CKKC-FC-CKV CKH-VFCRVCI CKKC-FC-KCV
A R N D C Q E G H I L K M F P S T W Y V 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 A
5 0 -2 -3 1 0 -2 0 -3 -2 2 -2 -3 -2 -1 -1 -3 -2 -3 R
6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 N
1 1 1 1 1
1 1
1 1 1 1 1 1 1 1 1 1
1
1
1
1 3 0 C 2 1 T 2 0 G 2 0 C 1 1 T 2 0 A 1 1 1 1 1 1 1 1 2 3 0 2 2 1 3 2 0 2 2 0 2 1 1 2 2 0 1 2 2 0 1 1 2 0 1 1 0 1 1 1 1 2 0 A 1 2 0 A 1 1 1 1 0 C
生物信息学中的序列比对算法原理与实践

生物信息学中的序列比对算法原理与实践序列比对是生物信息学中常用的基本技术之一,用于在生物学研究中比较两个或多个生物序列的相似性和差异性。
在分子生物学和基因组学等领域中,序列比对被广泛应用于基因分析、蛋白质结构预测、物种分类、进化分析以及新基因和功能区域的发现等重要任务。
本文将介绍序列比对算法的基本原理和常用实践技术。
序列比对算法的基本原理序列比对的目标是找到两个序列之间的匹配部分,并根据匹配的相似性和差异性进行评分。
序列比对算法的基本原理主要有两种方法:全局比对和局部比对。
全局比对算法(例如Needleman-Wunsch 算法)是一种通过将匹配、不匹配和间隙等操作分配给两个序列的每个字符来寻找最佳比对的方法。
它能够比较整个序列的相似性,但对于较长的序列来说,计算量较大,因此对于较短的序列和相似度较高的序列,全局比对更为合适。
局部比对算法(例如 Smith-Waterman 算法)则通过寻找两个序列中的最佳子序列来找到最佳比对。
该算法适用于较长的序列和不太相似的序列,因为它只关注相似的区域。
局部比对算法能够发现序列中的重复结构和片段,对于在序列之间插入或缺失元素的情况下非常有用。
序列比对算法的实践技术在实践应用中,为了处理大规模的序列数据并提高比对效率,还发展出了一些改进和优化的序列比对算法和技术。
1. 基于哈希表的算法:这种方法通过构建哈希表来加速相似性搜索。
算法将序列切分成较小的片段,并将每个片段哈希为独特的数字,然后根据相似性检索相关的哈希数字。
这种方法能够快速找到相似的序列片段,并进行比对和匹配。
2. 快速比对算法:这些算法通过减少比对的搜索空间或采用启发式的策略,来降低比对的计算复杂度。
例如,BLAST(Basic Local Alignment Search Tool)算法通过提取关键特征,如k-mer或频繁子序列,将序列比对问题转化为查找数据库中相似序列的问题。
3. 并行比对算法:随着计算机科学的发展,利用并行计算技术可以大幅提高比对效率。
生物信息学和计算生物学中的算法和模型

生物信息学和计算生物学中的算法和模型生物信息学和计算生物学是生物学领域的重要分支,致力于通过计算机科学的方法和技术来研究生物学中的各种问题。
从基因组学和蛋白质组学到系统生物学和进化生物学,生物信息学和计算生物学都发挥着重要的作用。
而算法和模型则是生物信息学和计算生物学的重要组成部分,为生物学研究提供了有效的理论和工具。
在本文中,将探讨生物信息学和计算生物学中的算法和模型的重要性和应用。
一、基于生物信息学的算法1.1 基因序列分析算法DNA的序列解码是生物信息学中最基本的问题之一。
基于生物学的算法广泛应用于基因序列的比对、组装和批量序列评估等领域。
基因序列分析算法涉及到与蛋白质互作、基因功能等生物学问题的关系。
基因组学技术的快速发展和大规模数据的产生,加速了基于生物信息学算法的研究进程。
1.2 蛋白质序列分析算法蛋白质是生命现象中不可或缺的一种物质,通过化学键形成了相对稳定的三维构型进行其特定的功能。
因此,分析蛋白质序列的方法与分析基因序列的方法有很多相似之处,但同时也存在很多不同之处。
蛋白质分析的目的是根据蛋白质的序列和三维结构,以推断其功能和保守区域。
研究者可以通过蛋白质序列分析算法和模型,预测蛋白质的结构和特性,以及通过相互作用和信号途径的分析,揭示蛋白质之间的关联性和影响性。
1.3 基于机器学习的算法机器学习是人工智能领域的一种重要技术,也是生物信息学中的重要方法之一。
生物信息学中的机器学习算法,例如基于神经网络的模型和基于支持向量机的学习算法,可以应用于生物学的数据分析中。
这些算法可以从数据中挖掘出结构,预测结果,并为生物学研究提供更加精确的计算分析。
二、基于生物信息学的模型2.1 基因调控模型基因调控模型是生物信息学中最为广泛应用的模型之一,因为大多数基因表达是在特定的环境条件下被调控的。
基因调控模型能够解析基因表达的模式和相应的信号途径,从而为生物学研究揭示更深层次的机制。
这些模型可以基于不同生物体在特定条件下的基因表达指标和外部条件,判断基因表达事件是否具有缓冲和分化的特性。
生物信息学中的序列比对算法使用方法解析

生物信息学中的序列比对算法使用方法解析序列比对在生物信息学中是一项重要的技术,用于寻找DNA、RNA或蛋白质序列之间的相似性和差异性。
它是理解生物学结构和功能的基石之一。
在本文中,我们将解析生物信息学中常用的序列比对算法的使用方法。
序列比对算法主要分为全局比对和局部比对。
全局比对用于比较完整的序列,而局部比对则更适用于在序列中查找相似区域。
在这两个主要类别中,有几种经典的序列比对算法,包括Pairwise Sequence Alignment、BLAST、Smith-Waterman算法和Needleman-Wunsch算法等。
首先,我们来看Pairwise Sequence Alignment(两两序列比对)算法。
这个算法是基本的序列比对方法,通过比较两个序列中的每一个碱基、氨基酸或核苷酸,并根据其相似性和差异性对它们进行排列。
Pairwise Sequence Alignment算法使用动态规划的思想,通过计算匹配、替代和插入/删除的分数,来确定两个序列的最佳匹配方案。
在生物信息学中,常用的实现包括Needleman-Wunsch算法和Smith-Waterman算法。
Needleman-Wunsch算法是一种全局比对算法,用于比较两个序列的整个长度。
它是通过填充一个二维矩阵来计算最佳匹配路径的。
算法的核心思想是,通过评估每个格子的分数,根据路径选择的最佳分数进行全局比对。
这个算法不仅可以计算序列的相似性,还可以计算每个位置的分数,从而获得两个序列的对应二面的对应关系。
Smith-Waterman算法是一种局部比对算法,用于寻找两个序列中的最佳匹配片段(子序列)。
它与Needleman-Wunsch算法的计算思路相同,但不同之处在于允许负分数,这使得算法能够确定具有高分数的局部匹配片段。
通过动态规划计算,Smith-Waterman算法可以寻找到两个序列中的相似片段,并生成比对的结果。
另一种常用的序列比对算法是基本本地搜索工具(BLAST)。
生物信息学中的基因组序列比对算法

生物信息学中的基因组序列比对算法生物信息学是一门研究生物数据的组织、分析和解释的学科,而基因组序列比对是生物信息学中的一项重要工作。
随着测序技术的飞速发展,已经可以获得大规模的基因组序列数据。
对这些海量数据进行比对,可以帮助科研人员更好地理解基因组的结构和功能,寻找与遗传疾病相关的基因变异,以及探索物种演化的关键基因。
基因组序列比对是指将已知的基因组序列与未知的基因组序列进行比较,找出相似的部分并进行对应的分析。
这个过程旨在寻找两个序列之间的共有特征,甚至找出它们之间的差异。
为了实现这个目标,生物信息学中发展了许多基因组序列比对算法。
本文将介绍几种常用的基因组序列比对算法和它们的特点。
1. Smith-Waterman算法:Smith-Waterman算法是最常用且最经典的基因组序列比对算法之一。
该算法的主要思想是通过动态规划的方式,找出两个序列之间的最优匹配。
它考虑了每个位置的匹配得分、插入得分和删除得分,并计算出匹配的最大得分。
然后,根据得分矩阵的反向路径,将匹配的结果进行回溯和确认。
Smith-Waterman算法的优点在于它能够找到最优的匹配结果,但缺点是计算复杂度较高,对于长序列的比对可能需要很长时间。
2. BLAST算法:BLAST(Basic Local Alignment Search Tool)算法是基因组序列比对中最常用的算法之一。
与Smith-Waterman算法相比,BLAST算法采用了一种快速比对的策略,以减少计算的时间复杂度。
BLAST算法首先将序列按照k-mer(由k个连续核苷酸组成的子串)进行分割,并将其转化为哈希表格式存储。
然后,在查询阶段,BLAST算法将查询序列的k-mer与目标序列的k-mer进行比较,从而找到相似的片段。
最后,根据相似片段的得分和位置信息,生成比对结果。
BLAST算法的优点是比较快速,但可能会因为基于k-mer的比对策略而丧失一些准确性。
生物信息学分析方法介绍PPT课件

目录
• 生物信息学概述 • 基因组学分析方法 • 转录组学分析方法 • 表观遗传学分析方法 • 蛋白质组学分析方法 • 生物信息学分析流程和方法比较
01
生物信息学概述
生物信息学的定义和重要性
定义
生物信息学是一门跨学科的学科,它利用计算机科学、数学和工程学的原理和 技术,对生物学数据进行分析、建模和解读,以揭示生命现象的本质和规律。
研究蛋白质的序列、结构 和功能,以及蛋白质相互 作用和蛋白质组表达调控 机制。
研究基因转录本的序列、 结构和表达水平,以及转 录调控机制。
研究基因表达的表观遗传 调控机制,如DNA甲基化 、组蛋白修饰等。
通过对患者基因组、蛋白 质组和转录组等数据的分 析,为个性化医疗和精准 医学提供支持。
02
基因组学分析方法
基因组注释
基因组注释是指对基因组序列中的各 个区域进行标记和描述的过程,包括 基因、转录单元、重复序列、调控元 件等。
注释信息可以通过数据库(如RefSeq、 GeneBank等)或注释软件(如GATK、 ANNOVAR等)获取。注释信息对于 理解基因组的生物学功能和进化关系 具有重要意义。
基因组变异检测
基因组变异检测是指检测基因组序列 中的变异位点,包括单核苷酸变异、 插入和缺失等。
VS
变异检测对于遗传疾病研究、进化生 物学和生物进化研究等领域具有重要 意义。常用的变异检测方法有SNP检 测、CNV检测等,它们基于不同的原 理和技术,具有不同的适用范围和精 度。
03
转录组学分析方法
RNA测序技术
利用生物信息学方法和算法,对 RNA测序数据进行基因融合检测, 寻找融合基因及其融合方式。
基因融合检测结果可以为研究肿 瘤等疾病提供重要线索,有助于 深入了解疾病发生发展机制。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
.精品课件.
14
生物序列相似性的比较
.精品课件.
15
•动机
在生物学的研究中,将未知序列同已知 序列进行比较分析已经成为一种强有力 的研究手段 ,生物学领域中绝大部分的 问题在计算机科学领域中主要体现为序 列或字符串的问题 。
.精品课件.
16
•序列联配问题的分类
如果两个序列具有足够的相似性, 则认为两者具有同源性。
.精品课件.
8
•蛋白质
由氨基酸依次链接形成在生物体中总共 有20种氨基酸。
蛋白有十分复杂的三维结构。其三维机 构决定了蛋白质的功能。
.精品课件.
9
•基 因
什么是基因?
DNA上具有特定功能的一个片断,负 责一种特定性状的表达。一般来讲, 一个基因只编码一个蛋白质。
.精品课件.
10
•基因组
-
序列相似性的比较 (两条序列的联配) 序列的分类 序列的排列 多序列的联配
.精品课件.
17
•两条序列联配问题的分类
全局联配(Global Alignment) 局部联配(Local Alignment) 空位处罚(Gap Penalty)
.精品课件.
18
•全局联配(1)-定义
定义1:两个任意的字符 x和y,(x,y)表示
表x和y比较时的分值。
(x,x)=2, (x,y)= (x,-)= (-,y)=-1
定可义以2用:序S列= sS1’…和sTn和’来T表=t示1…,tm其,中其:全局联配A (1) | S’ | = | T’ |; (2) 将S’和T’中的空字符除去后所得到的序 列分别为S和T;
联配A的分值Score为:
.精品课件.
19
•全局联配(2)-原始算法
输入:序列S和T,其中 | S | = | T | = n 输出:S和T的最优联配
for i=0 to n do for (S的所有的子序列A,其中| A | = i ) do for (T的所有的子序列B,其中| B | = i ) do
……
.精品课件.
0
0 -1 -2 -3 -4 -5
1 a -1 -1 1 0 -1 -2
2 c -2 1 0 0 -1 -2
3 g -3 0 0 -1 2 1
4 c -4 -1 -1 -1 1 1
5 t -5 -2 -2 1 0 3
6 g -6 -3 -3 0 3 2
.精品课件.
24
三种可能的最优联配序列:
1. S: a c g c t g T: - c – a t g t
转录:DNA链 → RNA链 信使RNA(mRNA),启动子。
翻译: mRNA上携带遗传信息在核糖体 中合成蛋白质的过程。
.精品课件.
7
•变异
进化过程中由于不正确的复制,使DNA 内容发生局部的改变。
变异的种类主要有以下三种: 替代(substitution) 插入或删除(insertion or deletion) indel 重排(rearrangement)
.精品课件.
4
•DNA(3)
DNA的双螺旋结构
碱基对之间的互补能力
.精品课件.
5
•DNA(4)
DNA的复制 在DNA解旋酶的作用 下两条链分离开,分 别作为一个模板,在 聚合酶的作用下合成 一条新链。
.精品课件.
6
•RNA、转录和翻译
RNA(核糖核酸):单链结构、尿嘧啶U代 替胸腺嘧啶T、位于细胞核和细胞质中。
2. S: a c g c t g T: - c a – t g t
3. S: - a c g c t g T: c a t g - t -
.精品课件.
25
•局部联配(1)
两条序列在一些局部的区域内具有 很高的相似度。
在生物学中局部联配比全局联配更 具有实际的意义。
两条DNA长序列,可能只在很小的区 域内(密码区)存在关系。
任何一条染色体上都带有许多基因,一 条高等生物的染色体上可能带有成千上 万个基因,一个细胞中的全部基因序列 及其间隔序列统称为genomes(基因组)。
.精品课件.
11
•DNA上的基因
基因
.精品课件.
12
•基因的编码
基因编码是一个逻辑的映射,表明存储 在DNA和mRNA中的基因信息决定什么 样的蛋白质序列。
不同家族的蛋白质往往具有功能和结 构上的相同的一些区域。
.精品课件.
26
•局部联配(2)
前提条件: V(i, 0) = 0; V(0, j) = 0;
20
•全局联配(3)
动态规划DP(Dynamic Programming) Smith-Waterman 算法
计算出两个序列的相似分值,存于一 个矩阵中。(相似度矩阵、DP矩阵)
根据此矩阵,按照动态规划的方法寻 找最优的联配序列。
.精品课件.
21
•全局联配(4)
前提条件
递归关系
.精品课件.
22
•全局联配(5)
在得到相似度矩阵后,通过动态规划回 溯(Traceback)的方法可获得序列的最 优联配序列 。
例: S = “a c g c t g”和T = “c a t g t”
(x,x)=2, (x,y)= (x,-)= (-,y)=-1
.精品课件.
23
j0 1 2 3 4 5
i
ca t g t
一种碱基
• 腺嘌呤(Adenine)
• 鸟嘌呤(Guanine)
• 胞嘧啶(Cytosine)
• 胸腺嘧啶(Thymine)
.精品课件.
3
•DNA(2)
碱基的配对原则 A(腺嘌呤)—T(胸腺嘧啶) C(鸟嘌呤)—G(胞嘧啶)
一个嘌呤基与一个嘧啶基通 过氢键联结成一个碱基对。
DNA分子的方向性 5'→3'
生物序列联配中的算法
张法
.精品课件.
1
•提 纲
背景知识 序列相似性的比较
两条序列的联配问题 多序列的联配问题 一些启发式的算法 生物序列联配中的并行算法
.精品课件.
2
•DNA(1) 脱氧核糖核酸
DNA的分子组成
核甘(nucleotides)
磷酸盐(phosphate)
糖(sugar)
每个碱基三元组称为一个密码子(codon) 碱基组成的三元组的排列共有43=64种,
而氨基酸共有20种类型,所以不同的密 码子可能表示同一种氨基酸。
.精品课件.
13
•带来的问题
序列排列问题 基因组的重排问题 蛋白质结构和功能的预测 基因(外显子、内含子)查找问题 序列装配(Sequence Assembly)问题