分离度不好,解决方法
提高分离系统的分离效果

提高分离系统的分离效果分离系统是化工、环保等领域中常用的设备之一,用于分离多种物质,使其得到纯化或者分别利用。
但是在应用过程中,分离系统分离效果可能会受到许多因素的影响,导致分离效果不理想,从而影响生产效率和经济效益。
因此,提高分离系统的分离效果就显得至关重要。
本文将介绍常见的提高分离系统分离效果的方法。
一、优化物料进出口位置和流量物料的进出口通常位于设备的顶部和底部,而流量的大小会影响到分离效果的好坏。
通过重新设计物料的进出口位置,可以使物料在设备内部的传递更加顺畅,流体的作用力更加平衡。
而通过调整流量大小,可以使物料更加均匀地分布在设备内部,从而避免物料在进出口周围的积聚,影响分离效果。
二、提高机械升温升压效果机械升温升压是一种常用的分离技术。
在进行机械升温升压时,可以通过提高压力和温度的大小,使物质中的组分达到不同的汽化温度和汽相分压,从而实现对物质的分离。
因此,提高机械升温升压效果可以使分离效果更加明显。
三、改进分离介质分离介质作为分离系统中的重要组成部分,其质量的好坏直接影响到分离效果。
在实际应用中,不同的介质适用于不同的物料分离,合理选择介质,可以明显提高分离效果。
同时,还可以通过优化介质的特性,例如表面活性剂、微观结构等,提高分离效果。
四、调整操作参数在进行分离操作时,涉及到的操作参数包括温度、压力、流量、反应物投入量等,这些操作参数直接影响到分离效果。
因此,通过调整操作参数,例如提高温度、加大流量等操作,可以使物料更加充分地混合、传递和反应,从而提高分离效果。
五、清洗设备分离设备的周期性清洗对于维护设备的正常运行和提高分离效果非常重要。
由于实际应用中,分离设备内部可能会出现不同程度的沉积物、结垢等,这些附着物不仅会影响分离效果,还会降低设备的使用寿命,增加维护成本。
因此,周期性清洗设备可以有效地去除这些附着物,保证设备的干净和高效运行。
六、应用增效剂增效剂通常是指一些具有增加分离效果的功能性物质,例如表面活性剂、络合剂等。
如何通过调节柱温提高物质分离度
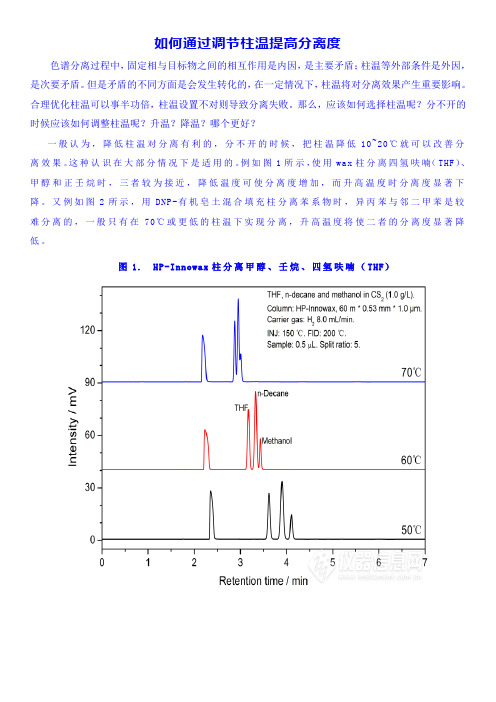
如何通过调节柱温提高分离度色谱分离过程中,固定相与目标物之间的相互作用是内因,是主要矛盾;柱温等外部条件是外因,是次要矛盾。
但是矛盾的不同方面是会发生转化的,在一定情况下,柱温将对分离效果产生重要影响。
合理优化柱温可以事半功倍,柱温设置不对则导致分离失败。
那么,应该如何选择柱温呢?分不开的时候应该如何调整柱温呢?升温?降温?哪个更好?一般认为,降低柱温对分离有利的,分不开的时候,把柱温降低10~20℃就可以改善分离效果。
这种认识在大部分情况下是适用的。
例如图1所示,使用wax柱分离四氢呋喃(THF)、甲醇和正壬烷时,三者较为接近,降低温度可使分离度增加,而升高温度时分离度显著下降。
又例如图2所示,用DNP-有机皂土混合填充柱分离苯系物时,异丙苯与邻二甲苯是较难分离的,一般只有在70℃或更低的柱温下实现分离,升高温度将使二者的分离度显著降低。
图 1. HP-Innowax柱分离甲醇、壬烷、四氢呋喃(THF)图 2. DNP与有机皂土混合填充柱分离邻二甲苯与异丙苯但是实际情况总是比较复杂的,降温并不总是有好处。
有些时候低温对分离反而不利,提高温度却能分离得更好。
这种情况其实并非个例。
例如图3所示,使用DB-1701柱分离正己酸与庚酸乙酯的时候,柱温低时二者十分接近难以分开,升高柱温反而使分离度增大。
又例如图4所示,使用DB-1701柱分离乙酸与正丁醇时也有类似情况。
这二者在柱温50℃时分离度比较低,降低柱温不仅无法改善分离度,反而使二者更加靠近。
反过来,增加柱温时二者的分离度却显著提高了。
图 3. DB-1701柱分离正己酸与庚酸乙酯图 4. DB-1701柱分离乙酸与正丁醇过犹不及,恪守中庸之道以上讨论表明,在气相色谱分析中,柱温的选择是比较复杂的,并不是简单的高温好还是低温好。
这时候很容易会想到儒家的中庸思想,认为太高或者太低都不好,适中才是最好的。
实际上这种变化规律也确实存在,而且在各种实验现象中有较为普遍的表现。
如何提高气相色谱仪的分离度

如何提高气相色谱仪的分离度?
有时在气相色谱仪的使用过程中,会发现样品复杂时容易分离不开的问题。
那么如何提高气相色谱仪的分离度呢?下面介绍几种常见的方法。
(1)、适当的增加柱长可以提高分离度。
(2)、减少样品的进样量(固体样品加大溶剂量降低浓度)。
(3)、提高进样水平防止造成两次进样。
(4)、降低载气的压力和流速。
(5)、降低色谱柱的温度使其分离更好。
(6)、提高汽化室的温度。
(7)、减少气路系统的死体积,比如色谱柱连接要插到位,不分流进样应选择不分流结构的汽化室。
(8)、毛细管色谱柱要分流,选择合适的分流比很重要。
综上,要提高气相色谱仪的分离度就要根据色谱峰型等来改变色谱仪的条件,或者样品的进样量等,最终达到分离好、出峰时间短的目的。
离子色谱中常用的改善分离度的方法

离子色谱中常用的改善分离度的方法1、稀释样品对组成复杂的样品,若待测离子对树脂亲合力相差颇大,就要作几次进样,并用不同浓度或强度的淋洗液或梯度淋洗。
对固定相亲合力差异较大的离子,增加分离度的最简单方法是稀释样品或作样品前处理。
例如盐水中SO2-4和Cl-的分离。
若直接进样,其色谱峰很宽而且拖尾,表明进样量已超过分离柱容量,在常用的分析阴离子的色谱条件下,30min之后Cl-的洗脱仍在继续。
在这种情况下,在未恢复稳定基线之前不能再进样。
若将样品稀释10倍之后再进样就可得到Cl-与痕量SO2-4之间的较好分离。
对阴离子分析推荐的最大进样量,一般为柱容量的30%,超过这个范围就会出现大的平头峰或肩峰2、改变分离和检测方式若待测离子对固定相亲合力相近或相同,样品稀释的效果常不令人满意。
对这种情况,除了选择适当的流动相之外,还应考虑选择适当的分离方式和检测方式。
例如,NO-3和ClO-3,由于它们的电荷数和离子半径相似,在阴离子交换分离柱上共淋洗。
但ClO-3的疏水性大于NO-3,在离子对色谱柱上就很容易分开。
又如NO-2与Cl-在阴离子交换分离柱上的保留时间相近,常见样品中Cl-的浓度又远大于NO-2,使分离更加困难,但NO-2有强的UV吸收,而Cl-则很弱,因此,应改用紫外作检测器测定NO-2,用电导检测Cl-,或将两种检测器串联,于一次进样同时检测Cl-与NO-2。
对高浓度强酸中有机酸的分析,若采用离子排斥,由于强酸不被保留,在死体积排除,将不干扰有机酸的分离。
3、样品前处理对高浓度基体中痕量离子的测定,例如海水中阴离子的测定,最好的方法是对样品作适当的前处理。
除去过量Cl-的前处理方法有:使样品通过Ag型前处理柱除去Cl-,或进样前加AgNO3到样品中沉淀Cl-;也可用阀切换技术,其方法是使样品中弱保留的组分和90%以上的Cl-进入废液,只让10%左右的Cl-和保留时间大于Cl-的组分进入分离柱进行分离。
离子色谱中常用的改善分离度方法

1、稀释样品对组成复杂的样品,若待测离子对树脂亲合力相差颇大,就要作几次进样,并用不同浓度或强度的淋洗液或梯度淋洗。
对固定相亲合力差异较大的离子,增加分离度的最简单方法是稀释样品或作样品前处理。
例如盐水中SO2-4和Cl-的分离。
若直接进样,其色谱峰很宽而且拖尾,表明进样量已超过分离柱容量,在常用的分析阴离子的色谱条件下,30min之后Cl-的洗脱仍在继续。
在这种情况下,在未恢复稳定基线之前不能再进样。
若将样品稀释10倍之后再进样就可得到Cl-与痕量SO2-4之间的较好分离。
对阴离子分析推荐的最大进样量,一般为柱容量的30%,超过这个范围就会出现大的平头峰或肩峰2、改变分离和检测方式若待测离子对固定相亲合力相近或相同,样品稀释的效果常不令人满意。
对这种情况,除了选择适当的流动相之外,还应考虑选择适当的分离方式和检测方式。
例如,NO-3和ClO-3,由于它们的电荷数和离子半径相似,在阴离子交换分离柱上共淋洗。
但ClO-3的疏水性大于NO-3,在离子对色谱柱上就很容易分开。
又如NO-2与Cl-在阴离子交换分离柱上的保留时间相近,常见样品中Cl-的浓度又远大于NO-2,使分离更加困难,但NO-2有强的UV吸收,而Cl-则很弱,因此,应改用紫外作检测器测定NO-2,用电导检测Cl-,或将两种检测器串联,于一次进样同时检测Cl-与NO-2。
对高浓度强酸中有机酸的分析,若采用离子排斥,由于强酸不被保留,在死体积排除,将不干扰有机酸的分离。
3、样品前处理对高浓度基体中痕量离子的测定,例如海水中阴离子的测定,最好的方法是对样品作适当的前处理。
除去过量Cl-的前处理方法有:使样品通过Ag+型前处理柱除去Cl-,或进样前加AgNO3到样品中沉淀Cl-;也可用阀切换技术,其方法是使样品中弱保留的组分和90%以上的Cl-进入废液,只让10%左右的Cl-和保留时间大于Cl-的组分进入分离柱进行分离。
对含有大的有机分子的样品,应于进样前除去有机物,较简单的方法是用Dionex的前处理柱OnGuard的RP或P柱或在线阀切换除去有机基体。
分子排阻分离度达不到

分子排阻分离度达不到摘要:一、分子排阻分离技术简介1.分子排阻分离技术的定义2.分子排阻分离技术的作用二、分子排阻分离度达不到的原因1.分子排阻柱选择不当2.样品的预处理不当3.操作条件不合适4.仪器故障三、解决方法1.选择合适的分子排阻柱2.优化样品预处理方法3.调整操作条件4.检查并维修仪器四、总结1.分子排阻分离度达不到的影响2.解决问题的意义正文:一、分子排阻分离技术简介分子排阻分离技术是一种利用样品中分子大小差异进行分离的方法,常用于生物大分子、聚合物、高分子溶液的分离。
该技术通过填充一定孔径的多孔材料,实现不同大小分子的筛选和分离。
二、分子排阻分离度达不到的原因1.分子排阻柱选择不当分子排阻柱的选择对分离效果至关重要。
如果选择的分子排阻柱孔径与样品中分子的大小不匹配,就可能导致分离度达不到预期效果。
2.样品的预处理不当样品的预处理是影响分子排阻分离效果的重要因素。
如果样品未进行充分搅拌、过滤或离心等处理,可能会导致大分子杂质进入分离柱,从而影响分离效果。
3.操作条件不合适操作条件包括流速、压力、温度等参数,对分子排阻分离效果具有重要影响。
如果操作条件设置不合理,可能导致分离度达不到预期效果。
4.仪器故障分子排阻分离仪器在使用过程中可能会出现故障,如柱塞损坏、柱床塌陷等,这些问题会影响分离效果。
三、解决方法1.选择合适的分子排阻柱针对具体样品,选择孔径合适、效果良好的分子排阻柱,确保分离效果达到预期。
2.优化样品预处理方法对样品进行充分搅拌、过滤或离心等预处理,降低大分子杂质对分离效果的影响。
3.调整操作条件根据具体样品和分子排阻柱的特点,合理设置流速、压力、温度等操作条件,确保分离效果达到预期。
4.检查并维修仪器定期检查分子排阻分离仪器,发现故障及时进行维修,确保仪器正常运行。
四、总结分子排阻分离度达不到预期效果会影响实验结果的准确性。
分子排阻分离度达不到

分子排阻分离度达不到1. 简介分子排阻分离是一种常用的生物化学技术,用于分离和纯化蛋白质和其他生物大分子。
然而,在实际操作中,有时会遇到分子排阻分离度达不到预期的情况。
本文将探讨分子排阻分离度不高的可能原因和解决方法。
2. 分子排阻分离度不高的可能原因2.1 样品质量问题样品质量是影响分子排阻分离度的关键因素之一。
如果样品含有杂质或降解产物,会影响到排阻柱的使用寿命和分离效果。
解决方法:在样品制备过程中,注意使用高质量的试剂和纯净的工作环境,避免杂质的污染。
此外,可以考虑使用降解产物较少的分离方法,如离子交换色谱。
2.2 分离柱选择不当不同的分子具有不同的分子大小和形状,因此需要选择适合的分离柱来实现有效的分离。
解决方法:在选择分离柱时,要考虑目标分子的分子大小和形状,以及分离柱的分离范围和分辨率。
如果选择不当,可以尝试更换其他类型的分离柱或调整分离柱的条件,如温度、流速等。
2.3 柱效问题柱效是指分离柱对目标分子的分离能力。
柱效不高会导致分子排阻分离度不高。
解决方法:在使用分离柱之前,要确保柱效良好。
可以通过检测柱效指标,如理论板数、高度当量等,来评估柱效的状态。
如果发现柱效不高,可以尝试进行柱效修复或更换新的分离柱。
2.4 分离条件不当分子排阻分离的条件包括流速、温度、缓冲液pH值等。
不当的分离条件会影响到分子的分离效果。
解决方法:在进行分子排阻分离时,要根据目标分子的性质和分离柱的要求来选择合适的分离条件。
可以通过调整流速、温度和缓冲液pH值等参数来优化分离效果。
3. 分子排阻分离度提高的方法3.1 样品预处理样品预处理是提高分子排阻分离度的重要步骤之一。
常用的样品预处理方法包括超滤、离心、沉淀等。
解决方法:根据样品的特点,选择合适的预处理方法。
例如,如果样品中含有大量的杂质,可以考虑使用超滤或沉淀的方法将杂质去除。
3.2 分离柱优化分离柱的优化是提高分子排阻分离度的关键。
可以通过调整分离柱的填料材料、长度、直径等参数来改善分离效果。
最全高效液相色谱法分离度不合格的原因及处理方法

高效液相色谱法分离度不合格的原因及处理方法
高效液相色谱法(HP1C)是一种常用的分析方法,用于分离和定量复杂混合物中的化合物。
分离度不合格可能有以下几个原因:
1.柱选择不当:不同的柱具有不同的分离性能和分离机理。
柱选择不当可能导致分离度不足。
建议根据样品性质选择合适的柱。
2.流动相问题:流动相的成分和流速对分离度有重要影响。
如果流动相成分不适合或者流速过高,可能会导致分离度不佳。
可以尝试调整流动相的成分和流速。
3.样品制备问题:样品的制备对分离度也有一定影响。
如果样品中有杂质或者未充分溶解,可能会影响分离度。
可以尝试优化样品制备方法。
4.柱温控制问题:柱温的控制对于一些温度敏感的化合物也非常重要。
如果温度控制不当,可能会影响分离度。
可以尝试优化柱温设置。
处理方法:
1.优化分析条件:根据分析物的性质和样品矩阵,优化柱的选择、
流动相的成分和流速等条件。
2.检查样品制备方法:确保样品制备充分、杂质减少,对于困难溶解的样品可以考虑使用适当的溶剂或提取方法。
3.检查仪器性能:检查液相色谱仪的性能,包括流速、温度、压力等参数是否正常。
4.质量控制:建立严格的质量控制标准,确保每一次分析都符合要求。
如果以上方法仍然不能解决问题,建议咨询有经验的分析人员或仪器供应商,以获得更专业的帮助。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
通常情况下首先会考虑调整流动相的比例您如果用的反相色谱应减少有机相,具体如何调整应参考3的原理,就是有机相减少,保留时间变长,如果原来是30%的有机相,现在减为20%,目标峰的保留时间会延长为原来的三倍;也有可能是柱子完了,峰分叉
原因主要有几种:
1 流动相不合适,更换流动相及重新摸配比。
2 可以通过调整流速达到更好的分离结果,如原来的1 ml/min,调到0.7ml/min,可能会有意想不到的效果。
3 还有一种可能是化合物部分氧化,用流动相按1:1稀释之后,再跑一次,可能分叉就没有了。
4 柱子可能有问题了,这个可以用标准品验证一下。
色谱柱不适合,或者流动相比例不好。
改变流动相比例或更换流动相。
某些情况下可以加入有机酸改变PH
呵呵,这个问题要根据具体情况来判断了,笼统的说有这样几种办法:
1,流动相改变。
可能改变流动相的PH,也可能改变流动相的配比,或用梯度来分离,还有可能用其它的流动相替换,这样根据样品结构及具体性质,还有实验结果来判断。
2,改变色谱柱。
可以用柱效更高的色谱柱来分离,也可能直接更换其它极性的色谱柱,也要看之前分离情况判断。
3,对样品进行前处理。
可能进行衍生,也可能进行萃取或其它方法。
4,如果最终还是无法分离,可以考虑诸如气相色谱等其它分析方法。
希望对你有所帮助。