?肺动脉高压诊断和治疗综述
BMJ综述:肺动脉高压的病因与治疗(二)
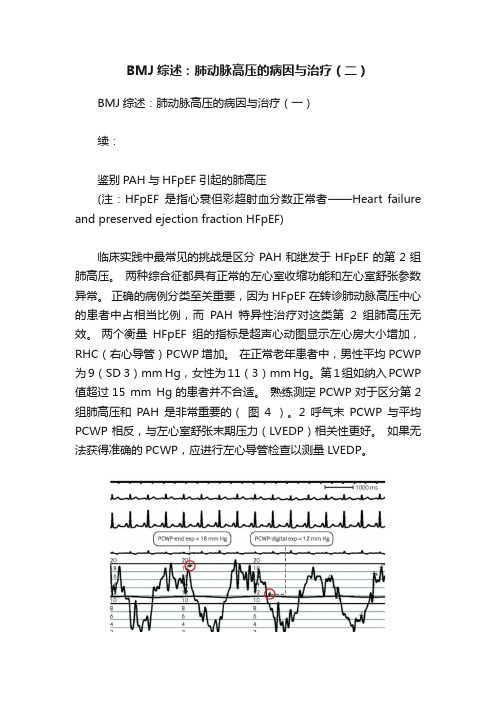
BMJ综述:肺动脉高压的病因与治疗(二)BMJ综述:肺动脉高压的病因与治疗(一)续:鉴别PAH与HFpEF引起的肺高压(注:HFpEF是指心衰但彩超射血分数正常者——Heart failure and preserved ejection fraction HFpEF)临床实践中最常见的挑战是区分PAH和继发于HFpEF的第2组肺高压。
两种综合征都具有正常的左心室收缩功能和左心室舒张参数异常。
正确的病例分类至关重要,因为HFpEF在转诊肺动脉高压中心的患者中占相当比例,而PAH特异性治疗对这类第2组肺高压无效。
两个衡量HFpEF组的指标是超声心动图显示左心房大小增加,RHC(右心导管)PCWP增加。
在正常老年患者中,男性平均PCWP 为9(SD 3)mm Hg,女性为11(3)mm Hg。
第1组如纳入PCWP 值超过15 mm Hg的患者并不合适。
熟练测定PCWP对于区分第2组肺高压和PAH是非常重要的(图4 )。
2呼气末PCWP与平均PCWP相反,与左心室舒张末期压力(LVEDP)相关性更好。
如果无法获得准确的PCWP,应进行左心导管检查以测量LVEDP。
图4 准确追踪测量肺毛细血管楔压(PCWP)的方法,以鉴别肺动脉高压和射血分数正常的心力衰竭所致的肺动脉高血压。
压力测量应在呼气末进行。
系统自行生成的平均压力会低估实际压力,注意避免。
Am Heart J 2012; 163:589-94临床与超声心动图模块流程可区分组第2组肺动脉高压和PAH (图5 )。
具有多种心血管合并症的老年患者因HFpEF而致肺高压的远比PAH多。
鉴于临床模块在区分继发于HFpEF和PAH的肺高压方面的准确率超过90%,超声心动图模块在识别PAH方面的特异性仅为59%。
为鉴别因于HFpEF或PAH所致的肺高压,可以尝试使用右心导管下的运动血液动力学评价和容量测试。
但是,这些技术尚未标准化,只能在经验丰富的中心使用。
肺动脉高压的诊断和治疗方法

肺动脉高压的诊断和治疗方法1.什么是肺动脉高压病?2.肺动脉高压做什么检查3.原发性肺动脉高压的治疗方法有哪些?什么是肺动脉高压病?肺动脉高压是各种原因引起的静息状态下右心导管测得的肺动脉平均压(meanpulmonaryarterialpressure,mPAP)≥25mmHg的一组临床病理生理综合征。
肺动脉高压可以作为一种疾病而独立存在,更常见的是很多疾病进展到一定阶段的病理生理表现。
由于肺血管重塑引起肺循环血流动力学改变,最终可导致右心衰竭,甚至死亡。
肺动脉高压是我国临床常见疾病,其致残和致死率颇高,也是严重危害患者身心健康,增加社会医疗负担的重大疾病。
这是一种极度恶性的疾病,七成多患者是年轻人,几乎每个人都知道癌症预后差,但没人知道肺动脉高压是一种极度恶性疾病,其愈后是灾难性的,可以说,这种病就是心血管疾病中的癌症。
肺动脉高压分为原发性(或特发性)和继发性两类。
其中原发性肺动脉高压是肺动脉的内皮肿瘤,相当于内皮细胞恶变后快速在肺动脉血管腔中生长并长满,所有静脉血因此都被肺动脉挡在一侧。
如无正确治疗,患者很快会死于难以纠正的右心衰竭。
肺动脉高压患者75%病人死于诊断后的5年内,症状出现后平均生存期为1.9年;有右心衰表现者,平均生存时间小于1年。
随着治疗手段的进步,患者生存时间在逐年增加。
及时早期诊断治疗可使20%病人的病情稳定,甚至痊愈。
这种疾病平均发病年龄是36岁,75%患者集中于20~40岁年龄段,还有15%患者年龄在20岁以下,几岁的孩子也会发病。
这是一种极易被误诊的疾病,临床应提高警惕。
肺动脉高压临床少见,临床症状缺乏特异性,如果接诊医师对肺动脉高压的诊断程序不清楚,不熟悉肺动脉高压的分类,易误诊为一般心脏病或者哮喘。
如临床医师注意到X线胸片有右心室肥大、肺动脉段突出而不能以常见的慢性阻塞性肺病、风心病、先心病、冠心病等疾病解释,即应想到本病可能,这时应进行超声心动图、心导管等检查检测肺动脉压力,并作有关系列检查。
肺动脉高压的诊断与治疗

肺动脉高压诊断和治疗进展北京协和医学院,中国医学科学院,阜外心血管病医院肺血管病诊治中心孙云娟 曾伟杰 朱锋 何建国**通迅作者:何建国,hejianguofw@gmail_com一. 肺动脉高压的定义和新概念1. 肺动脉高压(pulmonary hypertension,PH)的血流动力学定义为肺动脉平均压力(mean pulmonary arterial pressure,mPAP)在静息状态下>25mmHg,需用右心导管证实。
欧洲指南已明确指出:已发表的数据和健康人群资料不支持运动状态下mPAP>30mmHg作为PH的标准(见表1)。
2. 根据肺动脉楔压(pulmonary wedge pressure,PWP)是否大于15mmHg,分为毛细血管前肺动脉高压(PWP≤15mmHg)和毛细血管后肺动脉高压(PWP>15mmHg)。
(1)毛细血管前肺动脉高压(Pre-capillary PH)定义为平均肺动脉压≥25mmHg,且PWP ≤15mmHg,心输出量正常或降低;包括PH分类第1、3、4、5大类;(2)毛细血管和肺动脉高压(Post-capillary PH)定义为平均肺动脉压≥25mmHg,且PWP >15mmHg,心输出量正常或降低,包括第2大类。
3. 被动性PH(Passive PH)指跨肺压差≤12mmHg的PH。
4. 反应性PH(Reactive PH)指跨肺压差>12mmHg。
近年来,肺动脉高压领域的研究取得了令人瞩目的进展。
2009年美国心脏病学会和欧洲心脏病学会分别发表了肺动脉高压诊断和治疗专家共识/指南。
我国在肺动脉高压领域的研究也取得了可喜的进展。
本文将就肺动脉高压诊断和治疗方面的进展做一概述。
表1. 右心导管评估肺动脉高压的血流动力学标准*二. 肺动脉高压的分类欧洲《肺动脉高压诊断和治疗指南》新的临床分类沿用2008年在美国达纳波音特第四次表2. 最新的肺动脉高压临床分类(Dana Point,2008)1.1 特发性肺动脉高压1.2 可遗传性肺动脉高压 1.2.1 BMPR2 1.2.2 ALK1,endoglin(伴或不伴遗传性出血性毛细血管扩张症) 1.2.3 不明基因1.3 药物和毒物所致的肺动脉高压1.4 相关性肺动脉高压 1.4.1 结缔组织病 1.4.2 HIV感染 1.4.3 门脉高压 1.4.4 先天性心脏病 1.4.5 血吸虫病 1.4.6 慢性溶血性贫血1.5 新生儿持续性肺动脉高压1'. 肺静脉闭塞性疾病(PVOD)和/或肺毛细血管瘤病(PCH)2. 左心疾病所致的肺动脉高压2.1 收缩功能不全2.2 舒张功能不全2.3 瓣膜病3. 肺部疾病和/或低氧所致的肺动脉高压3.1 慢性阻塞性肺疾病3.2 间质性肺疾病3.3 其他伴有限制性和阻塞性混合型通气障碍的肺部疾病3.4 睡眠呼吸暂停3.5 肺泡低通气3.6 慢性高原缺氧3.7 发育异常4. 慢性血栓栓塞性肺动脉高压5. 原因不明和/或多种因素所致的肺动脉高压5.1 血液系统疾病:骨髓增生疾病,脾切除术5.2 系统性疾病,结节病,肺朗格汉斯细胞组织细胞增多症,淋巴管肌瘤病,多发性神经纤维瘤,血管炎5.3 代谢性疾病:糖原储积症,高雪氏病,甲状腺疾病5.4 其他:肿瘤性阻塞,纤维纵膈炎,透析的慢性肾衰竭肺动脉高压会议上提出的分类。
肺动脉高压的临床诊断及治疗研究

肺动脉高压的临床诊断及治疗研究一、概念介绍肺动脉高压(pulmonary arterial hypertension,PAH)是一种以肺动脉压力增高及肺血管阻力增加为主要特征的临床综合征,其病因多样,临床表现复杂。
PAH在欧美国家发病率为2-7/百万人口,而亚洲地区则较少见,发病率为0.37-1.1/百万人口,但中国普及度上升不容忽视。
二、病因分类PAH的病因较为复杂,可以按照其发生的基础疾病、遗传因素、自身免疫反应、血管异常等因素划分。
1、原发性肺动脉高压(idiopathic pulmonary arterial hypertension,IPAH)。
此种情况病因不明,约占PAH的40%。
2、家族性肺动脉高压(familial pulmonary arterial hypertension,FPAH)。
病因可能与细胞分裂周期调控和微血管内皮发育异常等遗传学因素有关,约占PAH的10%。
3、结缔组织病相关PAH(connective tissue disease-associated pulmonary arterial hypertension,CTD-PAH)。
如系统性红斑狼疮、硬皮病等,是PAH治疗中最常见的类型之一,约占PAH的30%左右。
4、肺栓塞后PAH(chronic thromboembolic pulmonary hypertension,CTEPH)。
当肺动脉血流受到阻碍,造成肺动脉血管壁的损伤和狭窄,以及肺循环系统的分流等,可引发CTEPH。
约占PAH的50%。
三、临床表现PAH多起病隐匿,早期症状不典型。
疾病发展到中期,则出现呼吸困难、乏力、胸痛等,使患者的日常生活和工作能力受到较大限制。
晚期症状严重而危及生命,包括心悸、晕厥以及腹水等症状,需及时救治。
四、诊断PAH诊断需要考虑病史、临床表现、心电图、胸部X线、心脏彩超、心导管和血气分析等多方面因素。
1、心导管检查。
可确定肺动脉平均压(mPAP)≥25 mmHg,波形呈圆顶型变化,同时肺血管阻力指数(PVR)>3 Wood units,除外左心病、肺病等疾病。
肺动脉高压的诊断和治疗

肺动脉高压的诊断和治疗肺动脉高压(PAH)是一种罕见且严重的疾病,主要表现为肺动脉血管壁增厚和肺血管阻力增加,导致心脏负荷加重、肺功能衰竭甚至死亡。
该病早期症状不典型,难以被发现,如果不及时诊治,疾病将不断恶化,治疗效果也会堪忧。
因此,及早诊断和治疗非常重要。
一、诊断目前,诊断PAH主要采用世界卫生组织(WHO)定义的标准,包括临床、肺功能和心脏超声三方面:1. 临床诊断:PAH主要症状包括呼吸困难、乏力、胸痛、心悸等,特别是运动后症状显著,如持续喘息、晕厥等。
另外,一些病人可能伴有手指端部疼痛或发绀等症状。
临床医生应通过详细询问病史和体格检查来确定是否存在PAH。
2. 肺功能检查:PAH病人肺功能常规检查往往正常,但因PAH导致肺小叶血管阻力增加,所以肺血管阻力增加的特征变化表现为肺血管压力升高,肺毛细血管楔压降低,肺血管反应性增加。
有时,肺功能测定可以提供对PAH严重程度的初步评估。
3. 心脏超声检查:是一种无创、易操作的检查方法,有助于诊断PAH。
超声检查可以直观评估心脏结构和功能,包括肺动脉压力、左心室和右心室大小等方面的细节。
此外,还可以评估心瓣膜功能以及心室大小和收缩功能,作为PAH的诊断依据。
二、治疗PAH的治疗旨在减轻症状、改善病情、延长生命,并提高生活质量。
目前,PAH的治疗方法主要包括药物治疗、物理治疗、外科手术治疗和心理治疗等多个方面。
1. 药物治疗:PAH的药物治疗主要采用下列几个类别的药物:(1)五磷酸酶抑制剂类(如西地那非):可刺激血管舒张,降低肺动脉压力。
(2)内皮素受体拮抗剂:可以减少血管收缩,降低肺血管阻力。
(3)前列腺素类(如腺苷酸酰化酶抑制剂):可以使血管扩张,降低肺动脉压力。
2. 物理治疗:包括氧疗、营养支持、呼吸道清理等。
氧疗可以改善肺功能,缓解病情。
营养支持可以改善病人的体力状况,提高营养状况。
呼吸道清理可以改善病人的呼吸道状况,减少发病风险。
3. 外科手术治疗:包括肺移植和手术治疗等。
【课题申报】肺动脉高压的诊断与治疗

肺动脉高压的诊断与治疗课题申报书一、课题背景和意义肺动脉高压(pulmonary arterial hypertension,PAH)是一种罕见但病情严重的血管疾病,其特征是肺动脉压力升高和肺血管阻力增加,导致肺循环血流受限,最终导致心功能不全。
该疾病的发病机制非常复杂,目前仍然没有根治的方法,对于肺动脉高压的早期诊断和有效治疗迫切需要进一步研究。
本研究拟以《肺动脉高压的诊断与治疗》为课题,旨在深入探索肺动脉高压的发病机制、风险因素、影响因素以及有效的诊断和治疗方法,为临床医生提供更好的指导和治疗方案,改善患者的生存质量和预后。
二、主要研究内容1. 分析肺动脉高压的病因和发病机制:通过收集大量的相关文献和病例资料,综述当前对于肺动脉高压病因和发病机制的研究成果,并对其进行总结和分析,明确肺动脉高压的病因学特点和发病机制。
2. 探究肺动脉高压的诊断方法及其价值:通过对肺动脉高压患者进行详细的病史采集、临床检查和辅助检查,分析不同方法在肺动脉高压的诊断中的准确性和价值,旨在提高肺动脉高压的早期诊断率。
3. 研究肺动脉高压的治疗方法及其疗效评价:根据肺动脉高压的病情严重程度和病因不同,采用不同的治疗方法,包括药物治疗、介入治疗和手术治疗等。
通过对患者进行长期随访和治疗效果评价,总结各种治疗方法的疗效与安全性,为临床医生提供更好的治疗方案和决策依据。
三、研究方法和进度安排1. 文献综述法:结合在线数据库和已发表的相关文献,对肺动脉高压的病因学特点、发病机制、诊断和治疗方法进行系统综述和分析。
2. 临床研究法:选取一定数量的肺动脉高压患者,进行病史采集、症状评估、体征检查和辅助检查,包括心电图、超声心动图、肺动脉压力测定等,进行分析和比较,评估不同诊断方法的准确性和价值。
3. 随访调查法:通过长期随访和疗效评价,观察和评估不同治疗方法的疗效、并发症发生率和生存率等,提供可靠的治疗参考依据。
预计本研究周期为三年。
肺动脉高压的病因与综合治疗

肺动脉高压的病因与综合治疗概述:肺动脉高压是指肺动脉血压持续升高,导致肺循环阻力增加的一种疾病。
其主要特点是肺动脉压力持续升高,导致右心室负荷过重,最严重时可发展为右心功能衰竭。
本文将探讨肺动脉高压的病因以及综合治疗措施。
一、病因分析1. 遗传因素遗传因素被认为是引起原发性肺动脉高压(pulmonary arterial hypertension, PAH)的主要原因。
约25%的PAH患者有家族遗传史,70%这种家族遗传史表现为自发突变。
2. 环境暴露长期接触到工业有害物质或药物也可能导致肺动脉高压。
比如:使用减肥药物苯巴比妥类和非法激素;暴露于有机溶剂、毒气和重金属等环境中。
3. 肺部疾病和血液系统异常某些肺部疾病,如慢性阻塞性肺疾病(COPD)和间质性肺纤维化,以及一些血液系统异常,如淋巴细胞增多症,都可导致肺动脉高压。
4. 先天心脏病先天性心脏病中的左向右分流型可引起肺循环血流量过大,导致肺动脉高压发生。
这种心脏结构缺陷可能是房间隔缺损、室间隔缺损或广泛的动静脉通道开放等。
5. 血栓栓塞急性大面积肺栓塞会造成肺血管阻力骤增,导致急性肺动脉高压。
长期慢性的血栓栓塞性肺动脉高压则是由多次反复的小面积肺栓塞所致。
二、综合治疗方案1. 药物治疗药物治疗是目前最常用的治疗方法之一。
主要包括以下几种药物:(1) 氨茶碱和茶碱类:通过扩张支气管和肺动脉使循环血流通畅。
(2) 血管紧张素转换酶抑制剂(ACEI)和血管紧张素受体阻滞剂(ARB):通过抑制血管紧张素转换酶的活性和阻断其受体,降低肺动脉压力。
(3) 钙离子拮抗剂:通过抑制钙离子进入肌细胞,减少平滑肌收缩,从而扩张肺动脉。
(4) 类脂洛地尔:可抑制磷酸二酰甘油途径和白三烯产生,具有降低大血栓球蛋白生成的作用。
2. 气瓶运动气瓶运动是通过参与有氧运动来改善患者的心肺功能。
适量的规律运动可以提高心肺功能、增加左/右心室间分流、促进外周吻合支的形成等。
肺动脉高压症的诊断与治疗

肺动脉高压症的诊断与治疗一、肺动脉高压症的概述肺动脉高压症(pulmonary arterial hypertension, PAH)是一种罕见但严重的心血管疾病,其特征为肺动脉压力升高。
PAH可以由多种原因引起,包括遗传因素、自身免疫性疾病、药物或物质暴露以及其他疾病的并发表现等。
PAH的诊断和治疗对于改善患者生活质量和预后至关重要。
二、肺动脉高压症的诊断1. 临床表现PAH在早期常无明显临床表现,随着病情进展,患者可出现呼吸困难、乏力、胸闷和心绞痛等不适感。
体格检查时可能发现颈静脉怒张、肝大、水肿等体征。
2. 影像学检查超声心动图是诊断PAH的首选非侵入性检查方法。
超声心动图可以评估右室功能、计算舒张末期右室压力和房间隔运动等指标。
3. 血液检查通过血液检查可以评估肺动脉高压的程度和病因。
常规检查包括肺功能试验、动脉血气分析以及相关生化指标的检测。
4. 心导管检查心导管检查是确认PAH诊断及评估疾病严重程度的金 standard,通过此项检查可以测量动脉压力、血流速度等参数。
三、肺动脉高压症的治疗目前,治疗PAH主要包括药物治疗、手术治疗和支持性护理。
1. 药物治疗目前已有多种药物被用于PAH的治疗。
常见的药物包括强心苷类药物、扩张剂、抑制剂等,其中扩张剂如硝酸盐类药物可降低肺动脉阻力,改善血液循环;而内皮素受体拮抗剂和PDE-5抑制剂可以减轻肺动脉收缩,并缓解血管壁细胞增殖。
2. 手术治疗对于一些适应证明确、有手术可能性以及无法通过药物治疗改善的患者,手术治疗是一个可行的选择。
肺动脉扩张术、肺移植和心脏移植等手术都可以被考虑。
3. 支持性护理支持性护理在PAH患者管理中起到重要作用。
合理控制液体摄入量、营养支持以及定期锻炼对于改善患者生活质量和功能状态有积极效果。
四、肺动脉高压症的预后PAH的预后取决于多种因素,包括年龄、诱因、基础疾病等。
未经治疗的PAH患者平均生存期仅为2-3年,而得到有效治疗的患者可显著延长生存时间。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
肺动脉高压诊断和治疗综述肺动脉高压诊断和治疗综述河北衡水哈励逊国际和平医院重症医学科高军译,王金荣校重症行者翻译组重要性:肺动脉高压(Pulmonaryarterial hypertension,PAH)是PH(pulmonaryhypertension,PH)的一个亚型,以肺动脉重塑为特征。
美国成人PAH的患病率约为10.6/100万。
如果未经治疗,PAH 会发展为右心衰甚至死亡。
观察:PAH是指平均肺动脉压>20mmHg,根据病因、病理生理学和治疗分为5个临床组。
PAH是其中的1组,通过插入右心导管确定血流动力学特征,显示平均肺动脉压>20mmHg,肺动脉楔压≤15mmHg,肺血管阻力≥3个Wood单位。
根据潜在病因,PAH进一步分为几个亚型,包括特发性PAH、遗传性PAH、药物和毒物相关性PAH、肺静脉阻塞性疾病、钙通道阻滞剂长期应答性PAH、新生儿持续性PH,以及与其他疾病相关性PAH包括结缔组织病、艾滋病和先天性心脏病。
早期症状非特异性,通常包括劳力性呼吸困难和疲劳。
目前已批准的PAH治疗药物包括增强一氧化氮-环鸟苷一磷酸生物途径的药物(西地那非、他达拉非或利奥西呱)、前列环素途径激动剂(依前列醇或曲前列环素)和内皮素途径拮抗剂(波生坦和安贝生坦)。
通过这些特异性治疗,PAH患者的5年生存率从1991年的34%提高到2015年的60%以上。
目前治疗包括针对一种以上生物途径的联合药物治疗,如一氧化氮-环鸟苷一磷酸和内皮素途径药物联合(如安贝生坦和他达拉非),与以前的传统单途径靶向治疗相比,其发病率和死亡率有明显改善。
结论和相关性:据估计,在美国每100万成年人中有10.6人患有PAH,并且如果未经治疗,通常会发展为右心衰甚至死亡。
以多种生物途径为靶点,以药物联合为一线治疗方法,可提高生存率。
肺动脉高压(Pulmonaryarterialhypertension,PAH)是一种危及生命的疾病,其特征是肺血管阻力增加,导致肺动脉压升高。
PAH 的症状是非特异性的,但通常包括劳力性呼吸困难和疲劳。
美国成年人PAH的患病率估计为10.6/100万。
如果未经治疗,PAH通常会发展为右室衰竭甚至死亡。
过去十年中,PAH的治疗显著改善了预后。
出现任何不明原因的劳力性呼吸困难都应考虑诊断PAH。
本综述总结了关于PAH诊断和治疗的当前证据。
一、方法从PubMed中检索1985年到2021年12月30日的英文文献,以“Pulmonaryarterialhypertension”为检索词,根据其与当前临床实践的相关性选择纳入的文章。
优先考虑随机临床试验、大型纵向观察研究和新发表文章。
在检索到文章的参考文献中搜索其他相关文章。
最后共纳入99篇文章,包括23项随机临床试验、3项meta分析和系统评价、36项观察性研究、16项基于注册登记的研究,以及21项临床实践指南或其他报告。
二、PH的定义和分类PAH是肺高压(pulmonaryhypertension,PH)的一个亚型,PH的临床分类对于理解PAH的诊断和治疗方法非常重要。
目前,PH 的定义是:静息状态下、仰卧位,插入右心导管测量平均肺动脉压大于20mmHg。
该定义不同于之前的25mmHg或更高的临界值,因为认识到与平均肺动脉压为20mmHg或更低的患者相比,平均肺动脉压为21-24mmHg的患者死亡率和住院率更高。
PH分为5组(图1A)。
第1组PH(PAH)定义为平均肺动脉压>20mmHg,肺动脉楔压≤15mmHg,肺血管阻力≥3个Wood单位;第2组,是与左心疾病相关的PH;第3组,与慢性阻塞性肺疾病或特发性肺纤维化和/或缺氧等肺疾病相关的PH;第4组疾病是肺动脉阻塞引起的PH,通常为慢性血栓栓塞性疾病;第5组是不明原因或多因素相关PH。
因此,尽管所有PAH(本综述主题)归类为PH,但只有第1组PH视为PAH。
三、PAH的病理生理学区分毛细血管前和毛细血管后异常导致的PH,对于理解PAH的病理生理学至关重要(图1B)。
PH是指肺动脉压升高。
正常情况下,右心室泵出的血液流经肺动脉和小动脉、肺毛细血管、肺小静脉和静脉,最终到达左心房。
当肺动脉压升高是由于肺毛细血管近端的肺动脉阻力增加所致时,存在毛细血管前型PH,并且表现为PAH。
PAH的病理生理学机制尚不清楚。
血管病理学包括动脉中膜和内膜重塑、丛状病变和弹性膜碎裂(图1C)。
血管重塑是指通过肥大或增生使血管壁增厚,导致肺血管阻力增加,随后平均肺动脉压增加。
血管收缩是PAH的重要组成部分,然而血管收缩空间是肺血管重构的始发环节(刺激事件)还是它的并发症,尚不确定。
最终毛细血管前动脉和小动脉被摧毁,导致PAH患者一氧化碳扩散能力(diffusingcapacity for carbonmonoxide,DLCO)降低。
因此,肺动脉重塑和血管收缩是PAH的治疗靶点。
最初,右心室通过增加收缩力和心室壁厚度来代偿后负荷的增加。
最终,右心室扩张并衰竭,如果不进行治疗,会由于右心衰而死亡。
四、PAH流行病学和患者特征PAH很少见。
法国注册登记研究(n=674,2002-2003年)报告年发病率约2.4例/100万人和15例/100万成人。
美国REVEAL注册登记研究(n=2525,2006-2007年)是一项涉及54个中心的多中心前瞻性队列研究,报告年发病率为2.0例/100万人和10.6例/100万成人。
不同注册登记研究的患者特征也不同。
与美国国立卫生研究院(NIH)1981年发起(n=187)的研究结果相比,REVEAL研究的PAH患者年龄更大(诊断时平均年龄分别为50.1岁[SD,14.4]和35岁[SD,15]),且女性患者更多(女性与男比例分别为4.07:1和1.7:1)。
目前尚不确定这些差异是否代表PAH患者特征的时间变化,或者是否与诊断治疗意识增强相关。
在进行针对性治疗之前,PAH确诊后的5年生存率为34%(NIH研究)。
而最近数据显示,1年生存率约为61%(REVEAL注册登记研究)和64%(FRENCH注册登记研究)。
五、PAH的亚组根据病理生理学、病因和对治疗的反应性,PAH进一步分为多个亚组。
特发性PAH(1.1组)特发性PAH以前称为原发性PAH,其符合PAH的血流动力学标准,但与其他疾病过程无关,如胶原性血管病或肝脏疾病。
根据REVEAL和FRENCH注册登记研究报道,特发性PAH的发病率分别为46%和39%。
遗传性PAH(1.2组)遗传性PAH(或家族性PAH)约占PAH患者的6%-10%。
骨形态发生蛋白受体-2基因(BMPR2)变异,占遗传性PAH的近75%,并且与常染色体显性/不完全外显性遗传模式相关。
当进行基因分型时,初始诊断为特发性PAH的患者约25%存在BMPR2变异,这就强调了对遗传性或特发性PAH患者进行遗传询问的必要性。
目前,在遗传性PAH患者中发现了大约15种其他变异体,包括遗传性出血性毛细血管扩张的变异。
与非遗传性特发性PAH相比,由BMPR2变异引起的遗传性PAH通常出现得更早(37岁VS.46岁),确诊时的血流动力学指标更严重,并且治疗反应更差。
药物和毒物诱发性PAH(1.3组)PAH与接触特殊药物和毒物有关,如甲基苯丙胺、达沙替尼或芬氟拉明。
一项针对2003-2015年住院患者的研究报告称,按照国际疾病分类编码,使用甲基苯丙胺的PAH相关住院率是非使用组的2倍多(分别是984.6例/100万和373.2例/100万;RR是2.64;95%CI2.18-3.2;P值<0.001)。
在90例甲基苯丙胺相关PAH和97例特发性PAH患者中,前者的病情更严重,进展更快(两人群5年和10年无事件生存率分别为47.2%和25%vs.64.5%和45.7%)。
达沙替尼是一种治疗肿瘤的酪氨酸激酶抑制剂,与PAH发生有关。
在FRENCH注册登记研究中2900名接受达沙替尼治疗患者,9名出现PH(诊断前平均治疗时间为31个月[8-48个月]),研究者向法国监管机构报告了另外4例与达沙替尼相关的PH病例。
因此,在有达沙替尼使用史的患者中,PH发生率最低估计为0.45%,而在不使用达沙替尼的人群中,为10-15例/100万。
达沙替尼相关性PAH通常会随着停药而消失。
在药物预警数据库中,36例达沙替尼相关PAH患者中有34例在停药后症状有所改善或缓解。
其他疾病相关性PAH(1.4组)结缔组织病相关性PAH(第1.4.1组)15%-25%的PAH与结缔组织疾病相关,最常见的是硬皮病,尽管也发生在系统性红斑狼疮、混合性结缔组织疾病、类风湿性关节炎和干燥综合征中。
硬皮病患者的PAH患病率为8%-14%。
与其他结缔组织疾病相关性PAH相比,硬皮病相关性PAH患者的预后更差。
与特发性PAH患者相比,心包积液患病率更高,6分钟步行距离更短,一氧化碳扩散能力(DLCO)更差。
存在PAH的硬皮病患者死亡率高于无PAH患者(3年生存率,94%VS.56%;n=546)。
世界PAH研讨会(WSPH)建议硬皮病患者每年进行超声心动图筛查,以促进PAH 早期诊断和治疗。
然而,没有临床试验表明这样做可以改善预后。
一项针对466名PAH高风险硬皮病患者的横断面国际研究发现,466名患者中有87名(19%)经右心导管插入术证实患有PAH。
HIV相关性PAH(1.4.2组)PAH是HIV感染的潜在并发症,在7658例HIV感染者中,PAH 的患病率为0.46%(95%CI,0.32%-0.64%),而在非HIV感染人群中,PAH的患病率为10-15/100万。
世界PAH研讨会建议对所有HIV 感染患者进行超声心动图评估,包括不明原因的呼吸困难或无症状HIV感染者,这些患者具有其他风险因素,如女性、静脉注射毒品或使用可卡因、或丙型肝炎感染。
然而并没有证明这种方法能改善HIV 患者预后。
门静脉高压相关性PAH(第1.4.3组)门静脉高压相关的PAH,可发生在肝硬化、非肝硬化性门静脉周围纤维化、门静脉血栓形成、肝静脉硬化和先天性门静脉循环异常所致门静脉高压患者。
当存在肝硬化时,门脉性肺高压患者的生存率更低。
在一项对1235名接受肝移植评估患者的前瞻性研究中,5%诊断为PAH。
门静脉高压与PAH之间的病生理联系尚不清楚。
据推测,在遗传易感个体中,肝功能障碍和门体分流,使肺血管床暴露于异常雌激素信号,因而产生不利影响。
实践指南建议对所有门静脉高压症患者进行超声心动图检查,以评估PH,尽管这种方法尚未证明能改善临床预后。
美国移植学会建议,将超声心动图筛查门脉性肺高压作为在肝移植前综合评估的一部分。
肝移植后,门脉性肺高压可能会改善或缓解。
对接受肝移植的门脉性肺高压患者(n=27)10年随访研究中,45%PH有所缓解,而55%需要进行肺血管扩张剂治疗。