药品补充申请分类
药品补充申请

药品填补申请药品填补申请(一)(省局审核,国度局审批部分)一.行政允许内容(一)持有新药证书的药品临盆企业申请该药品的同意文号.(二)应用药品商品名称.(三)增长中药的功效主治.自然药物顺应症或者化学药品.生物成品国内已有同意的顺应症.(四)变动用法用量或者变动实用人群规模但不转变给药门路.(五)变动药品规格.(六)变动药品处方中已有药用请求的辅料.(七)转变影响药品德量的临盆工艺.(八)修正药品注册尺度.(九)替代或减去国度药品尺度处方中的毒性药材或处于濒危状况的药材.(十)进口药品.国内临盆的打针剂.眼用制剂.气雾剂.粉雾剂.喷雾剂变动直接接触药品的包装材料或者容器;应用新型直接接触药品的包装材料或者容器.(十一)申请药品组合包装.(十二)新药的技巧让渡.(十三)修订或增长中药.自然药物解释书中药理毒理.临床实验.药代动力学等项目.(十四)进口药品在中国国内分包装.(十五)其他.五.申请材料应按请求报送以下相干材料,所有材料用A4纸制造,文字材料和表格用电脑打印,并按照下列次序分列:(一)申报材料封面即目次;(二)《药品填补申请表》(5份);注册申请人可自行在国度食物药品监视治理总局网站()高低载最新表格.(三)申报材料3套(按药品注册治理方法附件四请求供给)注:申报材料原件2套.复印件1套;复印件应该与原件完全一致,应该由原件复制并保持完全.清楚,用A4纸打印或复印药品填补申请(二)(省局审批,国度局存案部分)一.行政允许内容(一)转变国内药品临盆企业名称.(二)国内药品临盆企业内部转变药品临盆场地.(三)变动直接接触药品的包装材料或者容器(除上述第10事项外).(四)转变国内临盆药品的有用期.(五)转变进口药品制剂所用原料药的产地.(六)变动进口药品外不雅,但不转变药品尺度的.(七)依据国度药品尺度或者国度食物药品监视治理局的请求修正良口药品解释书.(八)填补完美进口药品解释书安然性内容.(九)按划定变动进口药品包装标签.(十)转变进口药品注册代理机构.(十一)其他.五.申请材料应按请求报送以下相干材料,所有材料用A4纸制造,文字材料和表格用电脑打印,并按照下列次序分列.(一)申报材料封面即目次;(二)《药品填补申请表》(4份);注册申请人可自行在国度食物药品监视治理总局网站()高低载最新表格.(三)申报材料1套(按药品注册治理方法附件四请求供给)(三)药品注册治理处对材料进行情势审查,不相符请求的应一次性告诉需补正的材料,相符请求的予以受理.请求进行现场核查和抽取样品的,在规准时限内完成现场考察和抽取样品,并通知省食物药品监视磨练研讨院进行样品磨练;不克不及受理的予以退审并解释来由.(四)下达《药品填补申请批件》,需样品磨练的,在收到样品磨练陈述后下达《药品填补申请批件》或《药品填补申请通知件》.(五)向国度食物药品监视治理总局上报存案.药品填补申请(三)(省局存案部分)一.行政允许内容(一)依据国度药品尺度或者国度食物药品监视治理局的请求修正国内临盆药品解释书.(二)填补完美国内临盆药品解释书安然性内容.(三)按划定变动国内临盆药品包装标签.(四)变动国内临盆药品的包装规格.(五)转变国内临盆药品制剂的原料药产地.(六)变动国内临盆药品外不雅,但不转变药品尺度的.(七)其他.五.申请材料应按请求报送以下相干材料,所有材料用A4纸制造,文字材料和表格用电脑打印,并按照下列次序分列.(一)申报材料封面即目次;(二)《药品填补申请表》(2份);注册申请人可自行在国度食物药品监视治理局网站()高低载最新表格.(三)申报材料1套(按药品注册治理方法附件四请求供给)。
药品注册分类5大类

药品注册分类5大类
一、一类药品:又称“特殊药品”,是用于治疗或预防特殊疾病以及有特殊用途的、
口服制剂、药物靶标复杂性或毒性较大的药品。
该类药品特殊性强,使用安全性较差,所
以药物注册历程要求高,批准理由也要十分慎重,属于国家控制的高危药品。
示例有:醋
酸咪唑、肝素钠粉剂、去氧核苷酸注射液和甲氨蝶呤等。
二、二类药品:又称“普通药品”,具有药物功能但不具有特殊功能或预防特殊病的
药物,是用于普通疾病的治疗、预防及日常养生的普通药品。
示例有:丙咪唑磷酸钠注射液、氢氯噻嗪片、氯化钾口服液、抗甲氧芬胶囊等。
三、三类药品:又称“简易药品”,具有较低的毒性和用药安全性的外用药品,如软
膏剂、凝胶、敷膏、油性外用液体等,主要用于缓解皮肤病症、肌肉骨骼外伤、局部感染
和烧伤疼痛等,常用于治疗局部病症。
示例有:多巴胺醋酸盐散料加强型冻干粉、地氯雌
醇外用凝胶、莫匹米诺磷酸钠喷雾剂等。
四、四类药品:又称“营养药品”,是一种补充营养素及有效成分,用于补充维生素、微量元素、氨基酸、氨基甘油酯、生育酚类成分及特质抗过敏物等,作用于增进机体疗效,增强抵抗力等,改善机体营养不良相关病症,综合应用于健康保健。
示例有:大豆异黄酮
胶囊、亚胺(阿斯巴甜)胶囊、钙镁磷片等。
五、五类药品:又称“中药材”,是以草本植物的全部或部分植物体(生药)或植物体
的制剂(中成药),通过特定的文献及历史证明,可用于预防和治疗疾病的复杂多冲精细混
合药物组合物。
其中一般分为生药和中成药,根据它们来源的不同,也可分为肉毒桿菌素类、原植物药类和药用动物类等。
示例有马鞭草、人参、当归、枸杞子、鱼腥草等。
医疗机构制剂补充申请申报资料项目及说明

附件2医疗机构制剂补充申请注册事项及申报资料要求一、补充申请分类(一)变更制剂名称(二)变更制剂规格(三)增加新的适应症或功能主治,但不改变给药途径或规范描述制剂功能主治(四)变更制剂处方中的辅料(五)变更制剂配制工艺(六)变更制剂有效期或贮藏条件(七)变更直接接触制剂的包装材料或者容器(八)变更制剂包装、标签样稿(九)根据国家或省药品监督管理局要求修改制剂说明书内容(十)补充完善制剂说明书安全性内容(十一)变更受托配制单位(十二)变更制剂配制地点(十三)变更制剂质量标准(十四)变更用法用量(十五)变更制剂包装规格(十六)变更医疗机构名称(十七)变更制剂外观,但不改变制剂标准(十八)其它二、补充申请申报资料项目1.制剂批准证明文件及其附件的复印件:主要是与申请事项有关的本制剂各种批准文件及附件。
2.证明性文件:申请人应当提供《医疗机构执业许可证》、《医疗机构制剂许可证》及其变更记录页;委托配制双方签订的委托配制合同、制剂配制单位《医疗机构制剂许可证》、《生产许可证》或《药品生产质量管理规范》认证证书复印件。
3.修订的制剂说明书样稿,并附详细修订说明。
4.修订的制剂包装标签样稿,并附详细修订说明。
5.配制工艺的研究资料及文献资料。
6.质量研究的试验资料及文献资料。
7.制剂的质量标准草案及起草说明。
8.制剂的稳定性试验资料。
9.三批样品的自检报告书。
10.辅料的来源及质量标准。
11.直接接触制剂的包装材料和容器的选择依据及质量标准。
12.主要药效学试验资料及文献资料。
13.毒理研究资料及文献资料。
14.临床试验资料。
15.支持该项变更的安全性研究资料及文献。
三、申报资料项目表注:*1.如有修改的应当提供。
*2.可提供毒理研究的试验资料或者文献资料。
*3.可提供文献资料。
四、注册事项说明及有关要求1.注册事项:第二项变更制剂规格,应当符合以下要求:所申请的规格应当符合科学、合理、必要的原则,所申请的规格应当根据制剂用法用量合理确定。
药品注册申请分类及申请基本要求

药品注册申请分类的意义
有利于规范药品研发
有利于保障公众用药安全
通过药品注册申请分类,可以明确不 同类型药品的研发要求和审批标准, 促进药品研发的规范化、标准化和科 学化。
通过科学合理的药品注册申请分类, 可以确保不同类型药品的安全性和有 效性得到充分评估和保障,从而保障 公众用药安全。
有利于提高审批效率
负责全国药品注册申请的监督管理工作,制定相关政策法规和标 准,对药品注册申请进行审批和监督检查。
省级药品监督管理部门
负责本行政区域内药品注册申请的受理、技术审查和审批等工作, 并配合国家药品监督管理部门进行监督检查。
市级药品监督管理部门
负责本行政区域内药品注册申请的现场核查等工作,并配合上级药 品监督管理部门进行监督检查。
审批程序的具体步骤
申请人向国家药品监督管理部门提交 药品注册申请,并按照规定提交相关 资料。
受理后,国家药品监督管理部门组织 开展技术审查,对药品的安全性、有 效性、质量可控性等方面进行全面评 估。
国家药品监督管理部门对申请资料进 行形式审查,符合要求的予以受理。
技术审查通过后,国家药品监督管理 部门作出是否批准的决定,并颁发药 品注册证书。
根据药品的研究阶段,将其分为临床 试验申请和生产申请。临床试验申请 是指申请开展临床试验的药品注册申 请;生产申请是指申请生产上市药品 的注册申请。
药品的风险程度
根据药品的风险程度,将其分为优先 审评审批、特别审批和快速审批等不 同类别。优先审评审批适用于具有明 显临床优势的创新药和有重大临床价 值的改良型新药;特别审批适用于未 在中国境内上市的境外新药;快速审 批适用于临床急需且符合国家食品药 品监督管理总局规定的其他情形。
关法规和标准的要求。
报国家食品药品监督管理局备案的药品补充申请分类及申报资料要求
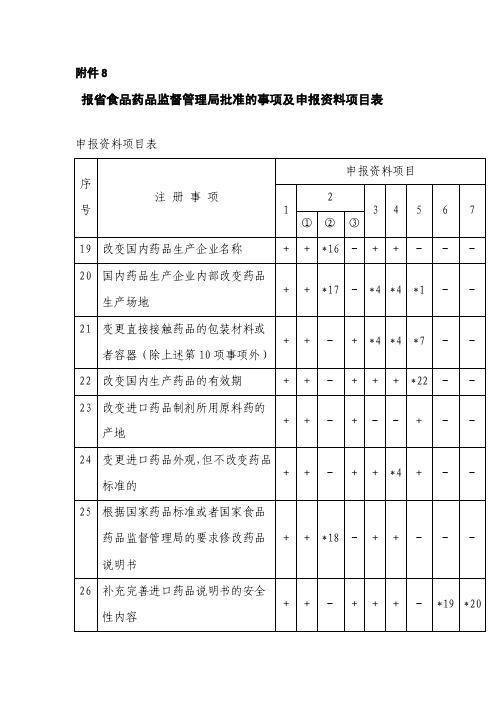
附件8报省食品药品监督管理局批准的事项及申报资料项目表申报资料项目表注:*1.仅提供连续3个批号的样品检验报告书。
*2.提供商标查询单。
*3.提供临床使用情况报告或文献。
*4.如有修改的应当提供。
*5.仅提供质量研究工作的试验资料及文献资料、药品标准草案及起草说明、连续3个批号的样品检验报告书。
*6有关毒性药材、处于濒危状态药材的证明文件,或者有关部门要求进行替代、减去的文件、证明。
*7.仅提供连续3个批号的样品检验报告书、药物稳定性研究的试验资料、直接接触药品的包装材料和容器的选择依据及质量标准。
*8.按照中药、天然药物、化学药品、生物制品注册分类中已在国外上市但尚未在国内上市销售的复方制剂的相应资料要求提供。
其中药学研究部分仅提供药物稳定性研究的试验资料、直接接触药品的包装材料和容器的选择依据及质量标准、连续3个批号的样品检验报告书。
*9.同时提交新药证书原件。
*10.提供技术转让有关各方签订的转让合同,原生产企业放弃生产的应当提供相应文件原件。
*11.国家食品药品监督管理局根据评价需要另行提出要求。
*12.提供包装厂所在国家或地区药品管理机构出具的该药品包装企业符合药品生产质量管理规范的证明文件。
*13.仅提供分包装工艺、药物稳定性研究的试验资料、直接接触药品的包装材料和容器的选择依据及质量标准、连续3个批号的样品检验报告书。
*14.提供进口药品分包装合同(含使用进口药品商标的授权)。
*15.仅提供分包装工艺、直接接触药品的包装材料和容器的选择依据及质量标准。
*16.提供有关管理机构同意更名的文件复印件,更名前与更名后的营业执照、《药品生产许可证》、药品生产质量管理规范认证证书等的复印件。
*17.提供有关管理机构同意药品生产企业变更生产场地的证明文件。
*18.提供新的国家药品标准或者国家食品药品监督管理局要求修改药品说明书的文件。
*19.可提供毒理研究的试验资料或者文献资料。
*20.可提供文献资料。
药品注册管理—进口药品、仿制药品、补充申请、再注册的管理

期、国内药品 审批,同时通知申请人。修改药品注
生产企业内部 改变药品生产
册标准的补充申请,必要时由药品检
场地等的补充 验所进行标准复核。
申请
(二)审批
按规定变更 药品包装标 签、根据国 家局的要求 修改说明书 的补充申请
报省级药品监督管理部门备案
由SFDA审批。
1.申请进 口药品分 包装要求
(5)除片剂、胶囊剂外 ,分包装的其他剂型应 当已在境外完成内包装
(4)分包装的药品应 与受托方所持有的许可 证和药品GMP证书上载
明的生产范围一致
(3)同一制药厂商的同 一品种应当由一个药品 生产企业分包装,分包 装期限一般不超过5年
(四)进口药品分包装的申报与审批
审批意见通知件
与
国家食品药品监督管理局
检验报告
审
同意 药
申请人
程
进口药品、仿制药品、补充 申请、再注册的管理
二、进口药品注册管理
( 一 ) 申请进口药品的要求
申请进口的药品必须获得境外制药厂商所在生产国家或者地区的
1 上市许可。未在生产国家或者地区获得上市许可,经国家食品药 品监督管理局确认该药品安全、有效而且临床需要的,可以批准 进口。
(二)进口药品再注册
《进口药品注册证》、《医药产品注册证》有效期5年。有效期届 满前6个月提出再注册申请。
(三)进口药品分包装的申报与审批
(1)申请分包装的药 品已经取得《进口药品 注册证》或《医药产品 注册证》
(2)该药品应当是中 国境内尚未生产的品种 ,或者虽有生产但是不 能满足临床需要的品种
进口药品的 补充申请
其中改变进口药品制剂所用原料药的产地、 变更进口药品外观但不改变药品标准、根据 国家药品标准或SFDA的要求修改说明书、补
药包材补充申请资料要求
药包材补充申请资料要求
一、药包材补充申请分类
(一)报国家食品药品监督管理局批准的补充申请事项:
1、变更药包材注册证所载明的“规格”项目。
2、变更药包材生产企业地址。
3、变更进口药包材注册证所载明的“公司名称”及“注册地址”。
4、变更药包材配方中原料产地。
5、变更药包材配方中的添加剂。
6、变更药包材生产工艺。
7、变更药包材注册标准。
(二)直接报国家食品药品监督管理局备案的补充申请事项:
8、变更进口药包材注册代理机构。
(三)由省级(食品)药品监督管理局审批,报国家食品药品监督管理局备案的补充申请事项:
9、变更国内药包材生产企业名称(含药包材生产企业地址变更名称)。
10、国内药包材生产企业内部变更药包材生产场地。
二、药包材补充申请申报资料项目
1、药包材批准证明文件复印件。
2、省级(食品)药品监督管理局对变更后的生产现场进行考核验收的报告。
3、国家食品药品监督管理局设置或者确定的检验机构出具的三批申报品种变更后的质量检验报告书原件。
4、国家食品药品监督管理局设置或者确定的检验机构出具的变更后的生产场地洁净室(区)洁净度检验报告书原件。
5、变更后的原料来源证明、执行的质量标准及其出厂质量检验报告书。
6、变更后的辅料来源证明、执行的质量标准及其安全用量的依据。
7、变更前后生产工艺对比研究资料。
8、变更注册标准的说明及变更前后的注册标准。
9、三批申报品种变更后的生产企业质量自检报告书原件。
10、采用变更后的申报品种包装的药品共同进行的稳定性试验研究资料(包含试。
药品补充申请
药品补充申请药品补充申请(一)(省局审核,国家局审批部分)一、行政许可内容(一)持有新药证书的药品生产企业申请该药品的批准文号。
(二)使用药品商品名称。
(三)增加中药的功能主治、天然药物适应症或者化学药品、生物制品国内已有批准的适应症。
(四)变更用法用量或者变更适用人群范围但不改变给药途径。
(五)变更药品规格。
(六)变更药品处方中已有药用要求的辅料。
(七)改变影响药品质量的生产工艺。
(八)修改药品注册标准.(九)替代或减去国家药品标准处方中的毒性药材或处于濒危状态的药材。
(十)进口药品、国内生产的注射剂、眼用制剂、气雾剂、粉雾剂、喷雾剂变更直接接触药品的包装材料或者容器;使用新型直接接触药品的包装材料或者容器.(十一)申请药品组合包装。
(十二)新药的技术转让。
(十三)修订或增加中药、天然药物说明书中药理毒理、临床试验、药代动力学等项目. (十四)进口药品在中国国内分包装。
(十五)其他。
五、申请材料应按要求报送以下相关材料,所有材料用A4纸制作,文字材料和表格用电脑打印,并按照下列顺序排列:(一)申报材料封面即目录;(二)《药品补充申请表》(5份);注册申请人可自行在国家食品药品监督管理总局网站(。
cn)上下载最新表格。
(三)申报资料3套(按药品注册管理办法附件四要求提供)注:申报资料原件2套、复印件1套;复印件应当与原件完全一致,应当由原件复制并保持完整、清晰,用A4纸打印或复印药品补充申请(二)(省局审批,国家局备案部分)一、行政许可内容(一)改变国内药品生产企业名称。
(二)国内药品生产企业内部改变药品生产场地。
(三)变更直接接触药品的包装材料或者容器(除上述第10事项外).(四)改变国内生产药品的有效期。
(五)改变进口药品制剂所用原料药的产地。
(六)变更进口药品外观,但不改变药品标准的。
(七)根据国家药品标准或者国家食品药品监督管理局的要求修改进口药品说明书。
(八)补充完善进口药品说明书安全性内容。
-药品补充申请的定义和类型
药品补充申请的定义和类型
(一)药品补充申请的定义
∙改变、增加或取消已批准的药品注册申请事项或内容需重新取得国家食品药品监督管理部门批准的注册申请。
∙新药技术转让,进口药品分包装,药品试行标准转正亦按照补充申请办理。
法规依据
《药品注册管理办法》第八条第四款补充申请,是指新药申请、已有国家标准的药品申请或者进口药品申请经批准后,改变、增加或取消原批准事项或者内容的注册申请。
(二)药品补充申请的类型
∙变更药品批准证明文件及其所附药品标准、药品说明书,标签载明事项的
∙改变生产工艺影响药品质量的相关事项
法规依据
《药品注册管理办法》第一百三十二条改变企业名称!按规定变更药品包装标签、根据国家食品药品监督管理局的要求修改说明书等的补充申请,由省、自治区、直辖市(食品)药品监督管理部门受理并审批,认为符合规定的,发给《药品补充申请批件》,并报送国家食品药品监督管理局备案;认为不符合规定的,发给《审批意见通知件》,并说明理由。
《药品注册管理办法》第一百三十三条修改药品注册标准、变更辅料、中药增加功能主治等的补充申请,由省、自治区、直辖市(食品)药品监督管理部门受理并提出审核意见后,报送国家食品药品监督管理局审批,同时通知申请人。
作者:发布时间:2008-9-24 14:54:37。
药品补充申请申报指南
药品补充申请申报指南药品补充申请申报指南⼀、报SDA批准的注册事项:1、持有新药证书的药品⽣产企业申请该药品的批准⽂号指新药研制单位获得新药证书时不具备该新药⽣产条件,并且没有转让给其他药品⽣产企业的,在具备相应⽣产条件以后,申请⽣产该新药。
资料要求:1.药品批准证明⽂件及其附件的复印件,同时提交新药证书原件2.证明性⽂件:GMP证书、⽣产许可证、营业执照3.修订的药品包装标签样稿(附详细修订⽐较说明)4.样品⾃检报告(3批)5.现场考察报告表6.省所检验报告(3批)注意事项:新药证书持有⼈应共同提出此项申请,即申请表中均应填写并共同盖章2、使⽤药品商品名称资料要求:1.药品批准证明⽂件及其附件的复印件2.证明性⽂件:GMP证书、⽣产许可证、营业执照、 \商标查询或注册证明3.修订的药品说明书样稿(附详细修订⽐较说明)4.修订的药品包装标签样稿(附详细修订⽐较说明)注意事项:1.药品商品名称仅适⽤于新化学药品、新⽣物制品。
2.中药不允许使⽤商品名。
3.不同规格使⽤⼀个商品名。
4.商标查询或受理注册证明的单位与申报⼈不⼀致,则需提供授权申报⼈使⽤该商标的合同(原件),同时在申请表的申请理由中注明。
5.商品名基本原则:商品名不能包含以下⽂字:有暗⽰疗效作⽤、有夸⼤或褒扬药品作⽤、有该药品通⽤名称、有⽣产单位名称、包含数字等。
6.新药拟使⽤商品名,应当由药品⽣产企业在申请新药注册时⼀并提出。
7.设⽴监测期的新药,在监测期内,申请⼈可以按照补充申请的要求申请增加商品名;监测期已过的药品不再批准增加商品名。
8.不设⽴监测期的新药,⾃批准⾸家注册后,2年内申请⼈可以按照补充申请的要求申请增加商品名;超过2年不再批准增加商品名。
9.新药保护期、过渡期已过的药品不再批准增加商品名。
3、增加药品新的适应症或者功能主治只能由药品⽣产企业提出申请。
资料要求:3.修订的药品说明书样稿(附详细修订⽐较说明)4.修订的药品包装标签样稿(附详细修订⽐较说明)5.药理毒理研究资料6.临床研究资料其药理毒理研究和临床研究应当按照下列进⾏:(1)增加新的适应症或者功能主治,需延长⽤药周期或者增加剂量者,应当提供主要药效学试验资料及⽂献资料、⼀般药理研究的试验资料或者⽂献资料、急性毒性试验资料或者⽂献资料、长期毒性试验资料或者⽂献资料,局部⽤药应当提供有关试验资料。