黄芩质量标准及检验操作规程
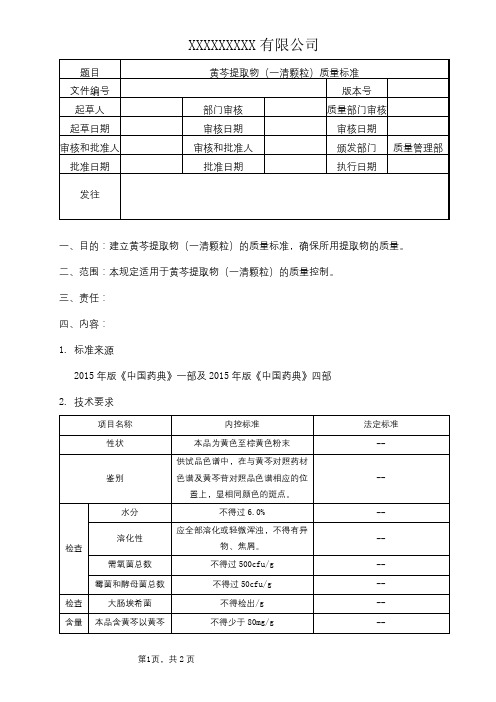
黄芩提取物(一清颗粒)质量标准
XXXXXXXXX有限公司
一、目的:建立黄芩提取物(一清颗粒)的质量标准,确保所用提取物的质量。
二、范围:本规定适用于黄芩提取物(一清颗粒)的质量控制。
三、责任:
四、内容:
1. 标准来源
2015年版《中国药典》一部及2015年版《中国药典》四部
2. 技术要求
3.贮存条件:密封,置阴凉干燥处。
4.相关标准操作规程:黄芩提取物(一清颗粒)检验操作规程(SOP-ZL-JG(ZJP)-015)、物料取样操作规程(SOP-ZL-QA-001)。
5.企业统一指定的物料名称:黄芩提取物(一清颗粒)。
6.内部使用的物料代码:无此项内容。
7.经批准的供应商:无此项内容。
8.包装形式:洁净内袋包装。
9.注意事项:无此项内容。
10.贮存期:24个月。
11.文件附件:共0份。
12.修订及变更历史:。
黄芩中药配方颗粒质量标准

黄芩中药配方颗粒质量标准
黄芩中药配方颗粒是一种中药制剂,主要以黄芩为主要原料,辅助药材可根据具体配方而定。
黄芩中药配方颗粒的质量标准应包括以下几个方面:
1. 外观特征:颗粒应为黄色或棕黄色,形状均匀,无结块、空心或明显异物。
2. 颗粒粒径:颗粒直径应符合标准要求,一般应为30目至80目。
3. 性状:颗粒应易溶于水,口感宜甘甜或微苦。
4. 含量测定:可根据具体配方确定黄芩和其他辅助药材的含量,并应符合国家药典或相应标准的要求。
5. 残留农药和重金属:应检测黄芩中农药残留和重金属含量,不得超过国家规定的安全标准。
6. 微生物限度:应检测细菌总数、霉菌和酵母菌数量,以及大肠杆菌、金黄色葡萄球菌等致病菌含量,并应符合相关国家标准。
7. 有关残留溶剂和杂质的限度,应根据具体配方以及相关药典或标准来确定。
以上是黄芩中药配方颗粒质量标准的一般要求,具体标准可根据实际情况和相关药典、标准进行制定。
黄芩的质量分析

黄芩的质量分析黄芩被称为中草药中的“不可多得的宝贝”,它的质量是影响用药效果的重要因素,因此质量分析是黄芩的重要研究内容。
黄芩的质量分析:1.分分析。
黄芩的成分包括:黄酮类、芳香类、多糖类、杀菌活性物质、金属及其他有机物。
它们是黄芩药效及药力的主要成分,要求黄芩的成分在各个组分中必须保持合理的比例,以保证其功效。
2.泽分析。
黄芩是一种植物,它的质量分析要通过色泽分析来确定黄芩植物的品种、新鲜程度及品质。
一般来说,新鲜的黄芩药材色泽黄绿相间,外观干燥,粒大、肥厚、柔软、伸条,具有明显的香气和清香。
3.分分析。
黄芩药材的水分含量是黄芩药效及药力的重要指标之一,它可以反映药材是新鲜的还是保存时间长的。
一般来说,新鲜的黄芩药材水分含量需要在15%以下,太高的水分含量将会影响黄芩的功效,因此要求质量的黄芩药材的水分含量一定要控制在15%以下。
4.质类分析。
杂质类分析是检查黄芩药材中杂质的指标。
一般来说,黄芩药材的杂质有外来物质、变质物质、腐朽物质等,这些杂质会影响黄芩的效力,可能对人体健康造成危害。
因此,要求黄芩药材中杂质的含量不得超过0.01%。
5.生物分析。
微生物分析可以帮助我们检测黄芩药材中的有害微生物,例如大肠杆菌、链霉菌、霉菌等,以确定黄芩药材是否安全可用。
要求黄芩药材中的有害微生物必须在国家规定的标准范围之内,以确保黄芩的质量。
6.金属分析。
重金属分析是检查黄芩药材中的重金属的指标,例如铅、镉、铜、汞等,它们是污染物,会严重影响黄芩的功效,甚至对人体健康造成危害。
因此,要求黄芩药材的重金属含量必须低于国家规定的标准,以确保黄芩的质量。
综上所述,黄芩的质量分析包括成分分析、色泽分析、水分分析、杂质分析、微生物分析和重金属分析等研究内容。
质量分析可以帮助我们检查黄芩药材的各项指标,以确保其功效及质量的可靠性,为人们提供安全有效的药物。
黄芩提取物质量标准及检验操作规程
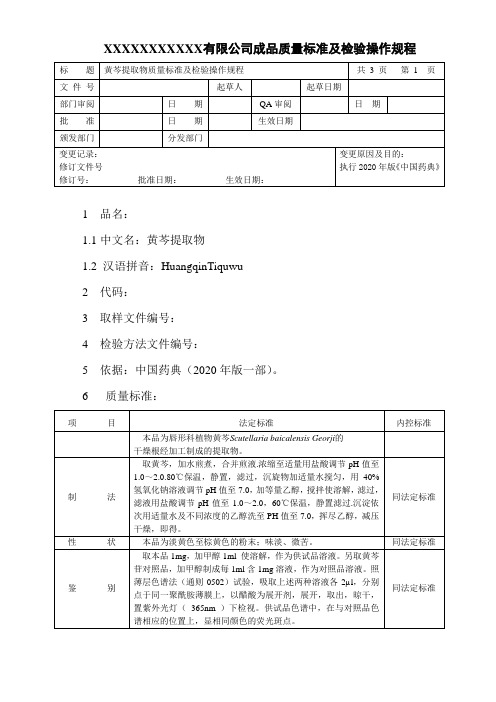
XXXXXXXXXXX有限公司成品质量标准及检验操作规程1 品名:1.1中文名:黄芩提取物1.2 汉语拼音:HuangqinTiquwu2 代码:3 取样文件编号:4 检验方法文件编号:5 依据:中国药典(2020年版一部)。
6 质量标准:7 检验操作规程:7.1 试药与试剂:甲醇、盐酸、氢氧化钠、乙醇、水、黄芩苷对照品、醋酸、磷酸。
7.2 仪器与用具:超声波清洗器、紫外光灯、高效液相色谱仪、电子天平、烘箱、马福炉、原子吸收。
7.3 性状:取成品,在自然光下目测色泽和形态、闻气尝味,并记录结果。
7.4 鉴别:取本品1mg,加甲醇1ml 使溶解,作为供试品溶液。
另取黄芩苷对照品,加甲醇制成每1ml含1mg溶液,作为对照品溶液。
照薄层色谱法(附录7)试验,吸取上述两种溶液各2µl,分别点于同一聚酰胺薄膜上,以醋酸为展开剂,展开,取出,晾干,置紫外光灯(365nm )下检视。
供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。
7.5 检查:7.5.1水分不得过5.0%(附录15 第二法)。
7.5.2炽灼残渣不得过0.8%(附录16)。
7.5.3重金属取炽灼残渣项下遗留的残渣,依法检查(附录13 第二法),不得过百万分之二十。
7.6含量测定:照高效液相色谱法(附录8)测定。
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇- 水- 磷酸(47:53:0.2 )为流动相;检测波长为280nm。
理论板数按黄芩苷峰计算应不低于2500。
对照品溶液的制备取黄芩苷对照品适量,精密称定,加甲醇制成每1ml 含60μg 的溶液,即得。
供试品溶液的制备取本品约10mg,精密称定,置25ml 量瓶中,加甲醇适量使溶解,再加甲醇至刻度,摇匀。
精密量取5ml,置25ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
黄芩检验标准操作规程
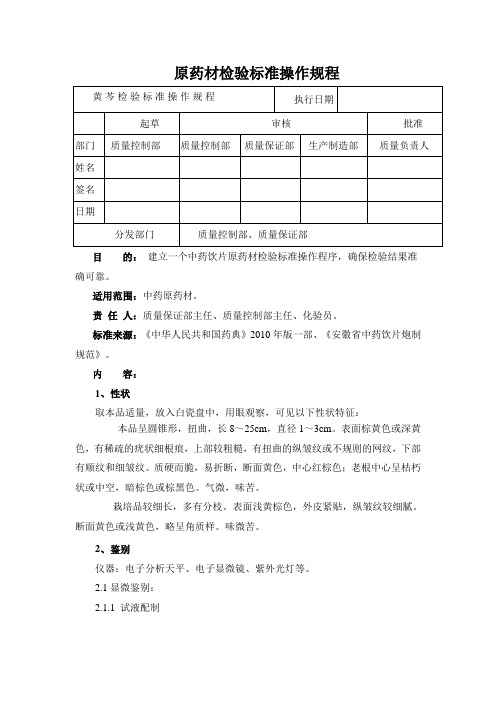
原药材检验标准操作规程目的:建立一个中药饮片原药材检验标准操作程序,确保检验结果准确可靠。
适用范围:中药原药材。
责任人:质量保证部主任、质量控制部主任、化验员。
标准来源:《中华人民共和国药典》2010年版一部、《安徽省中药饮片炮制规范》。
内容:1、性状取本品适量,放入白瓷盘中,用眼观察,可见以下性状特征:本品呈圆锥形,扭曲,长8~25cm,直径1~3cm。
表面棕黄色或深黄色,有稀疏的疣状细根痕,上部较粗糙,有扭曲的纵皱纹或不规则的网纹,下部有顺纹和细皱纹。
质硬而脆,易折断,断面黄色,中心红棕色;老根中心呈枯朽状或中空,暗棕色或棕黑色。
气微,味苦。
栽培品较细长,多有分枝。
表面浅黄棕色,外皮紧贴,纵皱纹较细腻。
断面黄色或浅黄色,略呈角质样。
味微苦。
2、鉴别仪器:电子分析天平、电子显微镜、紫外光灯等。
2.1显微鉴别:2.1.1 试液配制2.1.1.1 水合氯醛试液:取水合氯醛50克,加水15毫升与甘油10毫升使溶解,即得。
2.1.1.2 甘油醋酸试液:取甘油、醋酸及水各等份混匀,即得。
2.1.1.3 稀甘油:取甘油33毫升,加水稀释至100毫升,再加樟脑一小块或液化苯酚1滴,即得。
2.1.2 供试品制备2.1.2.1 取本品10g,研细后取少量粉末,置载玻片上,滴加水合氯醛搅拌均匀,置酒精灯上加热透化;加稀甘油数滴,搅拌均匀,分装2~3片,加盖玻片,即得。
2.1.2.2 取研细的粉末少量置载玻片上,加甘油醋酸试液,搅拌均匀,加盖玻片,即得。
2.1.2.3取研细后取少量粉末,置载玻片上,滴加水搅拌均匀,同时滴加少许稀甘油,加盖玻片,即得。
2.1.3 置显微镜下观察本品粉末黄色。
韧皮纤维单个散在或数个成束,梭形,长60~250µm,直径9~33µm,壁厚,孔沟细。
石细胞类圆形、类方形或长方形,壁较厚或甚厚。
木栓细胞棕黄色,多角形。
网纹导管多见,直径24~72µm。
木纤维多碎断,直径约12µm,有稀疏斜纹孔。
黄芩提取物质量标准及检验操作规程

黄芩提取物质量标准及检验操作规程需涵盖黄芩提取物质量标准及检验操作规程的全部内容;
一、黄芩提取物质量标准
1、外观:棕黄色淡黄色浓缩液。
2、比旋光度:20.0°~24.0°。
3、抗坏血酸(N-乙酰基氨基葡萄糖):≥1.8%。
4、Hesperidin:40.0~45.0%。
5、酸不溶性灰分:≤2%。
6、溶解度:1:1,8
7、PH值:4.5~5.8
8、活性蛋白酶活性:≤1000IU/ml。
9、乳球菌:≤1000CFU/ml。
10、大肠杆菌:≤100CFU/ml。
11、沙门氏菌:≤100CFU/ml。
12、霉菌:≤100CFU/ml。
1、黄芩提取物检验:取约3g样品,装入50ml用热弹性玻璃容器,加入到25ml的0.01mol/L浓硝酸,置摇床上振荡30min,再加入25ml甲醇,旋瓶至分离,用离心机15min,去除上清液,以清水洗涤残留液,将残留液中的活性物质分离出来,分别测定比旋光度、抗坏血酸(N-乙酰基
氨基葡萄糖)、Hesperidin、酸不溶性灰分、溶解度、PH值等,以符合规定的才算合格。
2、黄芩提取物微生物检验:取约5ml样品,用常温培养基灭菌,加入到玻璃盆中,然后再放入恒温器中培养,当检测时间到达一定时间后,用板株法测试乳球菌、大肠杆菌、沙门氏菌及霉菌等,符合规定的才算合格。
YL-11030黄芩原料检验操作规程
(W1-W0)×V0
浸出物% =×100%
W样×(1-水分)×V1
式中:
W0------- 蒸发皿的重量(g)。
W1-------- 浸出物与蒸发皿的重量(g)。
W样------- 样品的重量(g)。
V0-------加溶媒体积(ml)。
V1-------取续滤液的体积(ml)。
【含量测定】
【浸出物】照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用稀乙醇作溶剂,不得少于40.0%。
仪器与试剂:电热鼓风干燥箱、分析天平、蒸发皿、数显恒温水浴锅等。
方法:取供试品约4g,精密称定,置250~300ml的锥形瓶中,精密加水100ml,密塞,冷浸,前6小时内时时振摇,再静置18小时,用干燥滤器迅速滤过,精密量取续滤液20ml,置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量。除另有规定外,以干燥品计算供试品中水溶性浸出物的含量(%)。
m-------------------------样品的重量(g)。
f-------------------------稀释体积。
检验操作规程
题目:黄芩原药材检验操作规程
编号:TS-GC-YL-11020-02
制定人:
制定日期2020年 月 日
版本:1
页数:1/4
审核人:
审核日期2020年 月 日
颁发部门:质 量 部
批准人:
批准日期2020年 月 日
生效日期2020年 月 日
目的:规范黄芩原药材检验操作
范围:黄芩原药材检验
分发部门: 质量部、 化验室、生产部
照高效液相色谱法(通则0512)测定。本品按干燥品计算,含黄芩苷(C21H18O11)不得少于9.0%。
黄芩原料检验操作规程
制药GMP管理文件一.目的:为规范黄芩的检验标准和生产要求,特制定此标准。
二.适用范围:适用于中药材黄芩的质量检验。
三.责任者:质量检验员四.正文:检品名称:黄芩检验依据:《黄芩内控质量标准》检验仪器:显微镜三用紫外分析仪马弗炉干燥箱高效液相色谱仪操作内容:【性状】本品呈圆锥形,扭曲,长8~25cm,直径1~3cm。
表面棕黄色或深黄色,有稀疏的疣状细根痕,上部较粗糙,有扭曲的纵皱或不规则的网纹,下部有顺纹和细皱。
质硬而脆,易折断,断面黄色,中心红棕色;老根中心枯朽状或中空,呈暗棕色或棕黑色。
气微,味苦。
【鉴别】(1)本品粉末黄色。
韧皮纤维单个散在或数个成束,梭形,长60~ 250μm,直径9~33μm,壁厚,孔沟细。
石细胞类圆形、类方形或长方形,壁较厚或甚厚。
木栓细胞棕黄色,多角形。
网纹导管多见,直径24~72μm。
木纤维多碎断,直径约12μm,有稀疏斜纹孔。
淀粉粒甚多,单粒类球形,直径2~10μm,脐点明显,复粒由2~3分粒组成。
(2)取本品粉末lg,加乙酸乙酯-甲醇(3:1)的混合溶液30 ml,加热回流30分钟,放冷,滤过,滤液蒸干,残渣加甲醇5 ml使溶解,取上清液作为供试品溶液。
另取黄芩对照药材1克,同法制成对照药材溶液。
再取黄芩苷对照品,黄芩素对照品、汉黄芩素对照品,加甲醇制成每lml含lmg、0、5mg、0、5mg的对照品溶液。
照薄层色谱法试验,吸取供试品溶液、对照药材溶液各2μ1及上述三种溶液各1μ1,分别点于同一聚酰胺薄膜上,以甲苯-乙酸乙酯-甲醇-甲酸(10:3:1:2)为展开剂,预饱和30分钟,展开,取出,晾干,置紫外光灯(365纳米)下检视。
供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点;在与对照品色谱相应的位置上,显三个相同的暗色斑点。
【检查】总灰分不得过6.0%。
水分不得过12.0%。
【浸出物】照醇溶性浸出物测定法项下的热浸法测定,用稀乙醇作溶剂,不得少于40.0%。
黄芩含量测定实验报告
黄芩含量测定实验报告该实验是为了测定黄芩中黄芩素的含量。
实验操作步骤如下:1. 实验前准备:准备所需器材和试剂,包括黄芩样品、甲醇、1%硫酸乙醇溶液、氨基丁酸溶液。
2. 样品预处理:将黄芩样品粉碎成粉末状,称取适量黄芩样品,加入瓶中,并加入甲醇,在浸泡30分钟。
3. 萃取:将黄芩样品溶液进行超声萃取,即将瓶中的黄芩样品溶液放入超声浴中,进行震荡萃取。
4. 筛选:将超声萃取液离心,将上清液过滤,得到透明的黄芩提取液。
5. 过滤:将黄芩提取液过滤,过滤液收集起来备用。
6. 酸化:将过滤液加入1%硫酸乙醇溶液进行酸化,即加入适量硫酸乙醇溶液,并充分混合。
7. 萃取:将酸化液进行萃取,即加入适量氨基丁酸溶液,并进行震荡萃取。
8. 分液:将萃取液离心,得到上清液和沉淀物。
9. 沉淀增加:将沉淀物进行二次沉淀,即加入适量氨基丁酸溶液,并进行震荡沉淀。
10. 分液:将沉淀液离心,得到上清液和沉淀物。
11. 浓缩:将上清液进行浓缩,即将上清液放入水浴中,用蒸馏水浴进行浓缩。
12. 集合:将浓缩液转移到集合瓶中,并加入甲醇,溶解浓缩液。
13. 分液:将集合瓶中的液体进行离心,得到上清液。
14. 浓缩:将上清液进行再次浓缩,即将上清液放入水浴中,用蒸馏水浴进行浓缩。
15. 烘干:将浓缩液放入烘箱中进行烘干,直到得到稳定的干燥物。
16. 称量:将干燥物称取适量,并记录质量。
17. 计算:根据称取的干燥物质量,计算出黄芩中黄芩素的含量。
实验结果分析:根据实验得到的干燥物质量,可以计算出黄芩中黄芩素的含量。
要注意的是,该实验中的测定结果仅代表该批次黄芩样品的黄芩素含量,不能代表所有黄芩样品的含量。
实验中可能存在的误差主要有两个方面:1. 实验操作误差:包括称量误差、溶液制备误差等。
为了尽量减小这些误差,我们在实验中要注意仪器的准确使用和操作的规范性。
2. 样品本身的差异:由于黄芩样品可能存在不同的生长环境、采收时间等因素,导致不同批次的黄芩样品中黄芩素含量的差异。
黄芩中药配方颗粒质量标准
黄芩中药配方颗粒质量标准
黄芩中药配方颗粒的质量标准主要应包括以下内容:
1.来源与炮制:黄芩配方颗粒应来源于唇形科植物黄苓SCutellaria bacalens Georgi的干燥根,且应按照《中国药典》2010年版一部黄芩饮片的炮制方法进行加工制成。
2.性状:黄芩配方颗粒应为棕黄色至棕褐色颗粒,貝焦香气,味苦。
3.鉴别:应提供显微鉴别和理化鉴别方法。
以鉴别黄芩配方颗粒的真伪。
4.检查:应进行粒度、水分、灰分、酸不溶性灰分、重金属、农药残留等项目的检查,确保产品质量。
5.浸出物和含量测定:应规定浸出物和黄芩苷等有效成分的含量测定方法,以确保产品质量稳定、有效。
6.安全性评估:应对产品进行安全性评估,包括急性毒性、长期毒性、生殖毒性、致突变、致畸等试验,以确.保产品安全、无毒。
7.稳定性评估:应对产品进行稳定性评估。
包括加速试验和长期试验等,以确定产品的有效期。
8.其他指标:还应根据产品的实际情况制定其他相应的质量标准,如粒度分布、密度、润湿性等。
总的来说,黄芩中药配方颗粒的质显标准是一个综合性的标准,需要从多个方面来评估产品的质量。
制定:审核:批准:。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
XXXXXXX有限公司原料质量标准及检验操作规程
1 品名:
1.1 中文名:黄芩
1.2 汉语拼音:Huangqin
2 代码:
3 取样文件编号:
4 检验方法文件编号:
5 依据:《中国药典》(2020年版一部)。
6 质量标准:
7 检验操作规程:
7.1 试药与试剂:乙酸乙酯、甲醇、甲苯、甲酸、乙醇、黄芩对照药材、黄芩苷、黄芩素、汉黄芩素、磷酸。
7.2 仪器与用具:显微镜、三用紫外分析仪、恒温鼓风干燥箱、高效液相色谱仪、马福炉。
7.3 性状:取本品适量,自然光下目测色泽,嗅闻气味。
7.4 鉴别:
7.4.1 取本品制片置10×10显微镜下做显微观察。
7.4.2 取本品粉末1g,加乙酸乙酯-甲醇(3:1)的混合溶液30ml,加热回流30分钟,放冷,滤过,滤液蒸干,残渣加甲醇5ml使溶解,取上清液作为供试品溶液。
另取黄芩对照药材1g,同法制成对照药材溶液。
再取黄芩苷对照品,黄芩素对照品,汉黄芩素对照品,加甲醇分别制成每1ml含lmg、0.5mg、0.5mg的对照品溶液。
照薄层色谱法(附录7)试验,吸取供试品溶液、对照药材溶液各2μl及上述三种对照品溶液各1μl,分别点于同一聚酰胺薄膜上,以甲苯-乙酸乙酯-甲醇-甲酸(10 :3 :1 :2)为展开剂,预饱和30分钟,展开,取出,晾干,置紫外光灯(365nm)下检视。
供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点;在与对照品色谱相应的位置上,显三个相同的暗色斑点。
7.5 检查:
7.5.1水分:不得过12.0%(附录15第二法)。
7.5.2总灰分:不得过6.0%(附录17)。
7.5.3二氧化硫残留量照二氧化硫残留量测定法(附录58)测定,不得过150mg/kg。
7.6浸出物:照醇溶性浸出物测定法(附录19)项下的热浸法测定浸出物,用稀乙醇作溶剂,不得少于40.0%。
7.7含量测定照高效液相色谱法(附录8)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇-水-磷酸(47 :53 :0.2)为流动相;检测波长为280nm。
理论板数按黄芩苷峰计算应不低于2500。
对照品溶液的制备取在60℃减压干燥4小时的黄芩苷对照品适量,精密称定,加甲醇制成每lm|含60μg的溶液,即得。
供试品溶液的制备取本品中粉约0.3g,精密称定,加70%乙醇40ml,加热回流3小时,放冷,滤过,滤液置100ml量瓶中,用少量70%乙醇分次洗涤容器和残渣,洗液滤人同一量瓶中,加70%乙醇至刻度,摇匀。
精密量取lml,置10ml量瓶中,加甲醇至刻度,摇匀,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
本品按干燥品汁算,含黄芩苷(C21H18O11)不得少于9.0%。