药物作用新靶点与药-2010-09
新型药物靶点的发现与设计

新型药物靶点的发现与设计随着科学技术的不断进步和医疗需求的增大,新型药物的研发和设计显得尤为重要。
药物的研发过程中,寻找合适的药物靶点是关键一环。
本文将探讨新型药物靶点的发现与设计,介绍其中的方法和技术,以及未来的发展前景。
一、药物靶点的定义与重要性药物靶点是指药物与生物体内某一特定分子或结构发生相互作用并发挥药物效应的部位。
药物靶点的发现和选择对于药物研发至关重要。
一个好的药物靶点应该具备以下特点:与所治疗疾病密切相关、可调控性高、靶点的结构与功能了解程度较高等。
二、常用的药物靶点发现方法1. 基因组学方法基因组学方法通过对基因组数据的挖掘和分析,寻找与疾病相关的基因和蛋白质。
其中,基因芯片技术、序列比较和功能标记等方法可以帮助识别出与疾病相关的靶点。
2. 蛋白质组学方法蛋白质组学方法主要利用质谱技术和蛋白质相互作用的分析,寻找与疾病相关的蛋白质靶点。
通过分析蛋白质的组成、结构和功能等特性,可以筛选出潜在的药物靶点。
3. 化学生物学方法化学生物学方法主要是通过分子筛选和化合物库筛选等技术,寻找具有活性的化合物,进而发现药物靶点。
化学生物学方法的优势在于能够直接检测化合物与靶点的相互作用,缩小药物靶点的范围。
三、新型药物靶点的设计1. 确定药物的目标在设计新型药物靶点之前,需要明确所治疗的疾病类型和病理机制。
通过对与疾病相关的信号通路和分子机制的了解,可以选择合适的药物靶点。
2. 分子建模与计算机辅助药物设计分子建模技术可以通过计算机模拟和预测分子的结构和性质,以便更好地理解药物与靶点的相互作用。
计算机辅助药物设计则可以快速筛选出具有潜在活性的化合物。
3. 高通量筛选技术高通量筛选技术是一种高效的药物靶点设计方法,它可以通过快速筛选大量的化合物,找到与特定靶点相互作用的潜在药物。
四、新型药物靶点的发展前景随着基因组学、蛋白质组学和化学生物学等领域的发展,新型药物靶点的发现和设计在未来将取得更大的突破。
2009年美国FDA共批准26种新药上市

来源生物谷2010-1-26 10:59:57美国FDA2009年共批准26种新药上市赵晨光分享生物谷()讯:据统计美国食品药品监督管理局2009年共批准26种新药上市,而2008年FDA共批准25种新药上市。
在新批准上市的26种新药中生物技术类药物共有7种。
在药企方面,诺华制药以4种新药排在首位,强生旗下Centocor Ortho Biotech和葛兰素史克分别以两种新药紧随其后。
今年新药榜单上辉瑞和罗氏缺席的确令人吃惊。
以下是09年获批的26种新药:米那普伦盐酸盐 Savella (milnacipran HCl)适应症:纤维肌痛生产商:Forest Laboratories, Cypress Bioscience批准日期:2009年1月14日纤维肌痛是一种以全身性疼痛与疲劳为主的疾病。
根据美国风湿病学会估计大约有6百万美国人患有纤维肌痛。
虽然对于米那普伦盐酸盐(milnacipran Hcl,Savella)改善纤维肌痛症状的确切机制尚不明确,但一些研究者认为:某些脑部神经递质的异常可能是纤维肌痛的关键所在。
本品可能是通过对5-羟色胺与去甲肾上腺素(NE)再摄取的双重抑制(在体外对NE再摄取抑制的选择性更强)来改善纤维肌痛症状。
在美国进行的2项关键性III期临床研究中,共纳入2084例患者,其中1460例患者接受本品治疗,624例患者则使用安慰剂。
结果显示:与安慰剂相比,本品(剂量为100mg/天、200mg/天)能同时显著改善疼痛(视觉模拟评分)、患者整体评价(患者对变化的整体印象评分)与生理功能 (Short Form-36 总体生理健康)。
本品的安全性与耐受性良好。
最常见的不良反应为恶心。
其他常见不良反应包括便秘、潮热、多汗、呕吐、心悸、心率上升、口干与高血压。
本品所报道的大多数不良反应为轻微至中等程度。
Uloric (febuxostat)适应症:痛风生产商:Takeda批准日期:2009年2月16日Uloric (febuxostat)是美国FDA批准的近40年来首个用于治疗高尿酸症的痛风药物。
第3-4章2010.5试题(药学导论)答案

第3章---第4章单项选择题药物的解离度与生物活性的关系是(B )A. 增加解离度,有利于吸收,活性增加B. 合适的解离度,有最大的活性C. 增加解离度,离子浓度上升,活性增强D. 增加解离度,离子浓度下降,活性增强药物本身是有治疗效用的化学实体,在体内起作用后,经预料的和可控制的代谢作用,转变成无活性或无毒的化合物,此药物称为( D )A.硬药B.前药C.原药D.软药将氯霉素进行结构修饰制成氯霉素棕榈酸酯的目的是(A )A.改善药物的溶解性B.消除不适宜的制剂C. 提高药物对靶部位的选择性D.提高药物的稳定性不是发现先导化合物的途径是(C)A.天然产物中B.组合化学C.前体药物原理D.随机筛选研究开发新药,首先需要(C )A.研究构效关系B.进行剂型研究C.发现先导化合物D.进行药代动力学研究药物分子与受体之间的作用力包括(D)A.共价键和离子键B.共价键、离子键和氢键C.氢键、疏水作用和范德华力D.共价键、离子键、偶极作用、氢键、疏水作用和范德华力新药开发中属于药物化学研究范畴的是(A )A. 先导化合物的发现和结构优化B.剂型研究C. 临床研究D.临床前药动学研究由于药物与特定受体相互作用而产生某种药效的药物是(A )A. 结构特异性药物B. 前药C. 软药D. 结构非特异性药物药物作用过程的二个阶段中,药物与受体在靶组织的相互作用过程称为(C)A. 药剂相B. 药物动力相C. 药效相D. 药物代谢下列属于半合成药物的是( C )A. 青霉素GB. 青蒿素C. 阿莫西林D. 氯霉素下列属于天然药物的是( A )A. 青霉素GB. 氨苄西林C.头孢氨苄D. 苯唑西林下列属于从植物资源中得到的先导化合物是( A )A. 青蒿素GB. 青霉素G 阿司匹林C.D. 多西紫杉醇下列不属于提取药物的方法是(D)A.超临界流体萃取B.超声提取C.回流提取D. 溶剂分配法下列不属于天然药物化学的作用的是(B)A.探讨防病治病的药效物质基础B发现新靶点 C.改进传统剂型D. 扩大药物新资源银杏的药用部位属于哪一类化学成分(C )A.糖类B. 苷类C. 黄酮类D. 鞣质麻黄的有效成分属于哪一类化学成分(D )A.糖类B. 苷类C. 黄酮类D. 生物碱吗啡的化学成分类型为(D)A.糖类B. 苷类C. 黄酮类D. 生物碱多项选择题药物的亲脂性与生物活性的关系是( B c )A、降低亲脂性,一般可使药物作用时间缩短B、适度的亲脂性有最佳活性C、适当增强亲脂性,一般可使中枢作用药物作用增强D、降低亲脂性,有利吸收,活性增强E、增强亲水性,不利吸收,活性下降制成前药对药物进行化学结构修饰的目的是(ABCDE)A、提高生物利用度B、改善药物的吸收C、改善药物的溶解性D、提高药物的稳定性E、延长药物的作用时间下列可进行成酯修饰的药物是(AE)A、具有羧基的药物B、具有双键的药物C、具有卤素的药物D、具有氨基的药物E、具有羟基的药物先导物优化的一般方法是(BCDE)A、去除药效团B、结构拼合C、电子等排D、前体药物E、软药设计合理药物设计常用的靶点有(ABCDE )A、受体B、酶C、离子通道D、核酸E、基因属于溶剂提取方法的是(ABCE )A、煎煮法B、浸渍法C、渗滤法D、超声提取E、回流提取下列植物中含有小檗碱成分的是(ABD )A、黄连B、三棵松C、茶树菇D、古山龙E、杨柳树皮五、名词解释SAR :Structure-activity relationships,药物的化学结构与生物活性间的关系,通常称为构效关.NCE:New chemical entities, 具有一定生物活性的新型结构化合物,即为新化学实体.基本结构:通常具有相同药理作用类型的药物能与某一特定的受体或酶相结合,且它们在化学结构上往往具有某种相似性,在同类药物中化学结构相同的部分称为该类药物的基本结构。
2010年全球畅销药排名和变化(中文名)
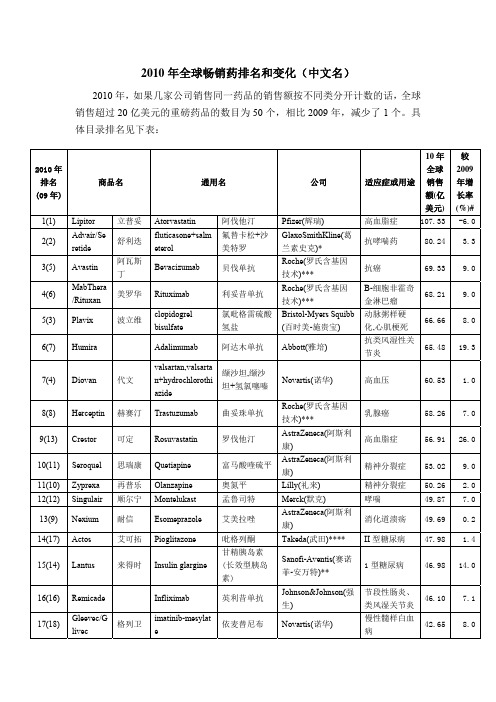
Losartan potassium
Telmisartan insulin lispro
他克莫司
Astellas Pharma(安 斯泰来)****
培美曲塞二钠 Lilly(礼来)
门冬胰岛素 (速效胰岛素)
人体胰岛素
氯沙坦钾,氯 沙坦钾+氢氯 噻嗪
替米沙坦
赖脯胰岛素
Novo Nordisk(诺和 诺德)***** Novo Nordisk(诺和 诺德)*****
品抗抑郁药文拉法辛(怡诺思,09 年排名第 29 位)、武田公司的兰索拉唑(达 克普隆,09 年排名第 38 位)、强生的阿法依泊汀(09 年排名第 45 位)、葛兰素 史克的伐昔洛韦(维德思,09 年排名第 50 位)和勃林格殷格翰治疗良性前列腺 增生的盐酸坦洛新(哈乐,09 年排名第 51 位)。
26.0
9.0 2.0 7.0 0.2 1.4
14.0
7.1
8.0
18(22) Spiriva
思力华 Tiotropium bromide
19(15) Lovenox
Enoxaparin
20(25) Prevnar
pneumococcal vaccine
21(23) Neulasta 22(21) Enbrel 23(28) Aricept
高血压 糖尿病
20.81 11.6 20.54 5.0
Risperidone
利培酮,利培 Johnson&Johnson(强 精神分裂症 酮长效注射剂 生)
20.27 -12.8
ezetimibe+simva 依折麦布+辛
sБайду номын сангаасatin
药物作用靶点发现的方法

药物作用靶点发现的方法药物作用靶点发现是药物研究的重要环节之一,它的目的是确定药物与生物体内的哪些分子相互作用,从而发现药物的作用靶点。
药物作用靶点发现的方法有很多种,下面将对其中的几种方法进行详细介绍。
1. 生物信息学方法生物信息学方法是一种基于计算机技术和生物学知识的方法,它可以通过对生物分子的序列、结构和功能等信息进行分析,预测药物与生物分子之间的相互作用。
其中,常用的生物信息学方法包括基于序列相似性的同源模拟、基于结构相似性的分子对接、基于功能相似性的基因表达谱分析等。
同源模拟是一种通过比对药物与已知蛋白质序列的相似性,预测药物与蛋白质之间的相互作用的方法。
这种方法可以通过比对药物与已知蛋白质序列的相似性,预测药物与蛋白质之间的相互作用。
同源模拟的优点是速度快、成本低,但缺点是预测结果不够准确。
分子对接是一种通过计算药物与蛋白质之间的相互作用能量,预测药物与蛋白质之间的相互作用的方法。
这种方法可以通过计算药物与蛋白质之间的相互作用能量,预测药物与蛋白质之间的相互作用。
分子对接的优点是预测结果较为准确,但缺点是计算成本较高。
基因表达谱分析是一种通过比较药物处理前后的基因表达谱,预测药物作用靶点的方法。
这种方法可以通过比较药物处理前后的基因表达谱,预测药物作用靶点。
基因表达谱分析的优点是可以发现新的作用靶点,但缺点是需要大量的实验数据支持。
2. 蛋白质组学方法蛋白质组学方法是一种通过对生物体内的蛋白质进行分析,发现药物作用靶点的方法。
其中,常用的蛋白质组学方法包括质谱分析、蛋白质芯片技术、蛋白质互作网络分析等。
质谱分析是一种通过对生物体内的蛋白质进行质谱分析,发现药物作用靶点的方法。
这种方法可以通过对生物体内的蛋白质进行质谱分析,发现药物作用靶点。
质谱分析的优点是可以发现新的作用靶点,但缺点是需要大量的实验数据支持。
蛋白质芯片技术是一种通过将蛋白质固定在芯片上,发现药物作用靶点的方法。
这种方法可以通过将蛋白质固定在芯片上,发现药物作用靶点。
新型药物靶点的发现与研究

新型药物靶点的发现与研究近年来,随着科技的不断进步和医学领域的不断探索,药物研究也日益成为研究的热点。
其中,药物靶点的发现与研究对于新药的开发尤为关键。
本文将探讨新型药物靶点的发现与研究的现状和未来发展。
一、背景介绍在药物研发的过程中,药物靶点是指药物分子与身体内的特定蛋白质、基因或细胞发生作用的位点。
药物通过与靶点结合来改变其结构或功能,从而实现治疗效果。
因此,发现与研究新型药物靶点被认为是新药研发的基石。
二、靶点发现的方法1. 基因组学方法基因组学方法通过研究整个基因组的功能与相互作用,识别出与特定疾病相关的基因或基因组区域,从而发现新的药物靶点。
目前,大规模基因测序技术的突破使得基因组学方法在药物研究中起到了关键作用。
2. 蛋白质组学方法蛋白质组学方法主要通过大规模分析蛋白质的表达和相互作用,发现与疾病相关的蛋白质,进而寻找药物靶点。
例如,蛋白质质谱技术可以帮助研究人员快速鉴定蛋白质样本中的蛋白质,揭示其功能和相互关系。
3. 化学基因组学方法化学基因组学方法将抗癌药物与大规模化合物库进行筛选,寻找具有特定抗癌作用的化合物,进而确定与其结合的蛋白质作为药物的靶点。
该方法可以帮助研究人员快速发现与疾病相关的新型药物靶点。
三、靶点研究的进展近年来,新型药物靶点的发现与研究取得了许多重要进展。
以癌症研究为例,许多新型药物靶点如细胞凋亡相关基因和修复基因等被发现并获得了临床应用。
1. 细胞凋亡相关基因细胞凋亡是机体去除异常细胞的一种重要途径。
研究表明,细胞凋亡相关基因如p53在多种癌症中发挥着重要作用。
因此,通过研究与细胞凋亡相关的新型药物靶点,可以为癌症治疗提供新的思路和方法。
2. 修复基因对于某些特定类型的癌症,肿瘤细胞的DNA修复能力异常活跃,从而导致药物治疗的失败。
研究人员发现抑制肿瘤细胞DNA修复机制的药物靶点,为癌症治疗提供了新的方向。
四、靶点研究的挑战与展望当前,虽然已经发现了许多重要的新型药物靶点,但仍然面临着许多挑战。
新型药物靶点发现与药物设计

新型药物靶点发现与药物设计随着科学技术的不断发展,药物研发领域也取得了显著的进展。
新型药物靶点的发现和药物设计成为了药物研发过程中的重要环节。
本文将介绍新型药物靶点的发现方法和药物设计的原理,以及相关领域的最新研究成果。
一、新型药物靶点的发现1.靶点的定义和重要性药物靶点是药物与生物体内相互作用的特定分子,通过与药物结合产生生物学效应。
靶点的选择和发现对于药物研发的成功至关重要。
不同疾病需要针对不同的靶点进行研究和设计。
2.基于基因组学的新型药物靶点发现基因组学的发展为新型药物靶点的发现提供了巨大的机会。
通过对基因组数据的挖掘和分析,可以发现一些与疾病发生和发展相关的基因,从而确定潜在的药物靶点。
3.蛋白质组学在药物靶点发现中的应用蛋白质组学的快速发展为药物靶点的发现提供了有力的手段。
通过蛋白质质谱分析和蛋白质互作网络的构建,可以筛选出与疾病相关的靶点,并进一步进行药物设计。
4.化学生物学方法的应用化学生物学方法结合高通量筛选技术可以高效地发现新型药物靶点。
通过荧光探针、亲和层析和酶活性测定等技术,可以筛选出与药物作用相关的分子靶点。
二、药物设计的原理1.药物设计的目标和原则药物设计的目标是合理设计出具有良好活性和选择性的药物,并降低其副作用。
药物设计的原则包括结构活性相关性、结构多样性和药物可控性等。
2.传统药物设计方法传统药物设计方法主要包括基于结构和基于药效模型的设计。
结构基础的设计是利用已知活性化合物的结构进行分析和优化,寻找结构相似的化合物作为候选药物。
药效模型的设计是根据已知的药效数据建立药效模型,预测潜在的候选药物。
3.计算机辅助药物设计计算机辅助药物设计利用计算机模拟的方法进行药物设计和筛选。
分子对接和量子力学计算等技术可以预测候选药物与靶点的相互作用,为药物研发提供理论和实验方向。
三、新型药物靶点发现与药物设计的最新研究成果1.基于人工智能的新型药物靶点发现人工智能在药物研发中发挥着重要作用。
药物新靶点的发现与研发

药物新靶点的发现与研发药物是维护人类健康的重要手段之一,而药物的研发过程又是一项复杂而艰辛的工作。
药物的研发包括药物发现和药物的临床开发,而药物发现又是药物研发过程中最基础、最重要的一环,也是决定药物能否成功应用于临床的关键因素之一。
药物新靶点的发现是药物发现过程中的重要一环,它指的是发现一些新的分子靶点或新的细胞信号通路,并通过对这些靶点或通路的研究和探索,发现新的治疗药物。
在过去的几十年中,药物研究主要鼓励发现新的化合物,因为这可以为新药的开发提供更多选择。
但是,在最近的研究中,越来越多的证据表明,通过发现新的药物靶点来寻找新药的策略可能更为有效。
药物新靶点的发现首先需要对疾病的原因和过程进行深入的研究。
这包括对疾病引起的分子或信号通路进行研究,探索其中的变异和调控机制,从而找到与该疾病相关的新的治疗靶点。
此外,研究人员还需要深入了解疾病的发展和进程,将疾病分类和分期,从而确定最适合的治疗方法。
当新的治疗靶点被发现后,就需要进行相关的药物研发工作。
药物研发工作包括药物的分子设计、合成和筛选。
药物分子设计是在了解药物靶点的结构和特性的基础上进行的,目的是寻找最优化的药物分子。
药物分子的合成是依据药物分子的结构和特性,通过化学合成的方法制备出理想的化合物。
药物的筛选则是对药物分子进行药理学和生物学评估,以确定是否具有治疗疾病的能力。
药物新靶点的发现和研发需要跨越多个学科和领域的知识,需要有很强的跨学科综合素质。
在药物研发过程中,化学家需要有深入的化学知识和实验技能,生物学家需要掌握各种细胞、动物模型,并熟练掌握相关的实验技术,药理学家需要了解药物的药理学作用和药物吸收、分布、代谢和排泄的规律。
此外,数据科学的应用也对药物研发过程中的数据处理、分析和可视化有很大帮助。
在对疾病治疗靶点的研究中,分子生物学、基因组学和生物信息学等新兴领域的进展也为药物研发带来巨大的机遇。
分子生物学提供了更多的靶点和信号通路供选择,基因组学和生物信息学则帮助药物研发人员更高效地筛选和评估药物靶点和药效。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
不可逆性抑制剂 以共价键与酶结合。此类抑制剂具有烷基化或 酰化(磷酰化)的功能,与酶形成稳定的共价键,
属活性试剂,可与组织和细胞中的氨基、巯基起作用。
抑制剂特点及要求
抑制剂必须到达作用部位即靶酶,且要维持一定 的浓度。影响这一要求的因素有排泄、代谢速率 以及为转运到靶酶所要求具有的恰当的亲脂性与 亲水性比率。 抑制剂应有特异性,即其作用仅限于靶酶。因为, 如果与其他酶或维持细胞正常代谢必需的细胞成 分反应,在临床上将带来不良的甚至是危险的副 作用。
当前热点
以端粒酶为靶点的药物设计 端粒酶是由小分子RNA和蛋白质组成的一种特 殊核糖核蛋白逆转录酶。端粒酶被认为是一种广 泛的肿瘤标志,端粒酶活化参与细胞的癌变过程 已得到肯定。端粒酶通过延长端粒而维持肿瘤细 胞的持续增殖能力。许多细胞通过端粒酶的表达 下调或酶裂解而退出细胞周期。在正常生长条件 下,抑制端粒酶活性,肿瘤来源的细胞将继续分 裂并伴随端粒的缩短,最终处于静息状态或死亡。
G-蛋白偶联受体
G-蛋白偶联受体(G-protein coupling receptor)其 介导的信号传导系统 GPCR由单一肽链形成的7个α 螺旋反复穿透细胞膜,并与异源三聚体G蛋白偶联形 成跨膜区段,也称为7次跨膜受体 当外来化学信号作用于相应的靶细胞膜表面时,能与 膜表面的G蛋白偶联型受体分子中第7个跨膜螺旋能 够识别并特异性结合。受体因结合了特异化学信号而 被激活后,将进而作用于膜中的G-蛋白质。 如腺苷受体、肾上腺素受体、多巴胺受体、组胺受体、 阿片受体等
R H3 C N N O R H3 C N N H H N NH O N NH O RCH2NH2 H3C H3C R N H N NH
O
H3C
N H NH O CH H R
O RCH=NH H2 O
H 3C
RCHO
NH3
当前热点
抗癌新靶点——MAP4K3 在肿瘤研究所工作的科学家们最近发现了位于 AP4K3上的一个抗癌新靶点。MAP4K3是MAP(有 丝分裂原激活性蛋白)激酶中的一员:属于丝/苏氨 酸特异性蛋白激酶,可对外源性刺激(有丝分裂 原)产生反应,并调节诸如基因的转录、有丝分 裂、细胞分化、存活和调亡等多种细胞活动。具 有“开启”增殖启动子mTOR的作用。
酶抑制剂的设计原理与方法
1.定向活性部位抑制剂
2.基于机理的抑制剂
定向活性部位抑制剂
特点:识别基团--与酶可逆结合的部分,结构
上相似于底物的某些片断,其作用是将抑制剂引 入到酶的活性部位。 反应活性基团多为亲电试剂。
定向活性部位不可逆抑制剂
糜蛋白酶的活性部位与TPCK的结合状况
His57 Asp102 COOSer195 O C CH O N H HN CH H C C
β-淀粉样前体蛋白APP
阻止 代谢
加入 前体 物
黄嘌呤氧化酶(OX)抑制剂(可逆)
OH N N N N H 次黄嘌呤 HO OX N N 黄嘌呤 OH N N 别嘌醇 N H OX HO N N N N OH N OX N H HO N N 尿酸 OH N OH N H
OH N
体内的嘌呤代谢紊乱或摄入嘌呤过多 都会导致体内的嘌呤含量过高,从而 使得尿酸生成过剩,导致其在关节等 处沉积,最终形成痛风
OH
Bu N
HO OH
脱氧野尻霉素的 N-丁基衍生物
OH
血管紧张素转化酶抑制剂(可逆)
血管紧张素酶原 → 血管紧张素Ⅰ→ 血管紧张素Ⅱ→ 血管收缩 → 外周阻力增加 → 血压升高
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Protein 1 2 3 4 5 6 7 8 9 10 11 12 13 肾素 Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu 1 2 3 4 5 6 7 8 9 10 血管紧张素Ⅰ Asp-Arg-Val-Tyr-Ile-His-Pro-Phe 1 2 3 4 5 6 7 8 血管紧张素Ⅱ
前
言
药物作用靶点(d rug target)是指具有重要生理或病理功能, 能够与药物分子相结合并产生药理作用的生物大分子(基因 位点、受体、酶、离子通道、核酸等)及其特有的结构域
目前已经发现的药物作用靶点约有500个.研究表明, 蛋白质、核酸、酶、受体等生物大分子不仅是生命的 基础物质,有些也是药物的作用靶点。现有药物中, 以受体为作用靶点的药物超过50%,是最主要和最重 要的作用靶点;以酶为作用靶点的药物占20%之多, 特别是酶抑制剂,在临床用药中具有特殊地位;以离 子通道为作用靶点的药物约占6%;以核酸为作用靶点 的药物仅占3%;其余近20%药物的作用靶点尚待研究 发现。
Receptors
Most receptors are proteins that traverse the cell membrane with a binding site on the extra-cellular region. Binding of a chemical messenger causes the receptor to change shape, initiating a process that results in a message being received by the cell. The messenger dose not undergo any reaction and departs unchanged, allowing the receptor to reform its origin shape. Binding site of the receptors is equivalent of an enzyme`s active site, but has no catalytic activity.
药物作用靶点与 药物合理设计
生物医学工程研究室:顾月清
本章提纲
以酶为靶点的药物设计
1
2
以受体为靶点的药物设计
3
抗肿瘤新靶点及药物设计
前
言
药靶是药物在体内的作用结合位点,包括基因 位点、受体、酶、离子通道、多糖、核酸等生物 大分子。迄今已发现作为治疗药物靶点的总数约 500个,其中受体靶点占绝大多数(50%)。药靶 的寻找是新药研究与开发的关键,合理化药物设 计(rational drug design)可以依据生命科学研 究中所揭示的包括酶、受体、离子通道、核酸等 潜在的药物作用靶位,或其内源性配体以及天然 底物的化学结构特征来设计药物分子,以发现选 择性作用于靶点的新药。
二、以受体为靶点的药物设计
受体(receptor)是存在于细胞膜表面或胞浆内 能与特异性配基相结合并产生生物学效应的大 分子物质,也定义为:受体是能与激动剂高度 选择性结合,并随之发生特异性效应的生物大 分子或大分子复合物)。 1878年 Langley 首先提出受体概念 20世纪30年代, Clark提出了配基与受体相互作 用的占领学说
•阿尔茨海默病(Alzheimer`s disease, AD)早老性痴呆
早老性痴呆
1 抑制β-和γ-分泌酶
皮质神 经元减少
皮下神经元系 统功能衰退 胆碱乙酰转移 酶活性降低
淀粉样蛋 白团块
神经纤 维缠结
胆碱摄取 能力下降
小胶质细胞激活 Aβ(40 or 42) β-和γ-分泌酶切割 胆碱 酯酶 抑制 剂 激活 胆碱 受体 等 增加 递质 的释 放
细胞质膜受体
根据功能不同又可将细胞质膜受体分为 通道性受体 细胞质膜受体 G-蛋白偶联受体 催化性受体 水溶性配基如多肽、生物胺难以通过细胞膜,其 受体一般为质膜受体。
细胞质膜受体
① 通道性受体(channel-linked receptor) 通道性受体(channel-linked receptor)是质膜的跨 膜蛋白,一般由4~5个肽链组成,每一肽链反复多 次穿透细胞膜组成跨膜离子通道,5个亚基中的每个 亚基沿离子通道中心轴排列为准五重对称的结构, 且每个亚基均含有4段跨膜结构。 功能是通过增强特定离子的瞬间透过性控制神经系 统中反应最快的突触效应,介导可兴奋性信号的快 速传导 。如乙酰胆碱受体、兴奋性氨基酸受体、抑 制性氨基酸受体等
受体的分类
按照国际药理学联合会(IUPHAC)和药物分类委员 会(NC-IUPHAR)的建议,将受体分为四大类,即 受体分子四大家族,G-蛋白偶联受体、通道性受体、 酶性单链跨膜受体(又称催化性受体)和配基依赖 的转录因子受体等。 根据细胞中受体存在的位置不同,按照受体的蛋白 质结构和功能,特别是与细胞膜的结合功能及信号 转导特征,将受体分为: 细胞质膜受体 G-蛋白偶联受体、通道性受体、催 化性受体。 胞内受体:配基依赖的转录因子受体
H 奥昔嘌醇
黄嘌呤氧化酶是嘌呤分解代谢过程中的一种很重要的酶,催化由黄嘌呤形 成尿酸以及次黄嘌呤形成黄嘌呤的氧化过程。它是一种含钼的黄素蛋白,也
能催化醛类的氧化反应,与多种疾病如痛风、肝炎等密切相关
该酶抑制剂如5-甲酰尿嘧啶、重金属离子、咪唑等
αβ-葡糖苷酶抑制剂
降血糖药阿卡波糖就是αβ葡糖苷酶抑制剂。 从天然产物中得到的氮杂 糖类化合物,作为糖苷酶 抑制剂防止糖蛋白合成中 形成异常N-连接的多糖链 结构、改变病毒糖蛋白的 多糖结构而影响病毒的感 染性,如脱氧野尻霉素的 N-丁基衍生物具有强的抗 HIV作用
一、以酶为靶点的药物设计
酶(Enzyme)绝大多数化学本质为蛋白,
是生物体内最重要的功能大分子。以酶为靶 点的药物设计(20%)有两种途径,即通过物 理的或化合的方式与酶结合,对酶的活性起 到增强或抑制效果,而在药物开发及临床应 用上,酶抑制剂类药物占绝大多数。
降脂药物的潜在作用靶点——酰基辅酶A-胆固醇酰基转移酶