链霉菌总DNA提取
改良的CTAB法提取霉菌DNA

前言:今天终于把一株黑曲霉DNA提取出来了,由于实验室条件简陋,购买试剂盒不划算,只好发挥实验室一切能用到的方法:冰冻、水煮、微波、超声波、溶菌酶解,等等效果都不佳,无奈,这次结合几种方法在一起,虽然有点麻烦,但是效果还可以,用来扩增16S没有一点问题,提取霉菌DNA只是雕虫小技,在此献丑了。
改良的CTAB法提取霉菌DNA1、PDA液体培养基,32℃过夜培养菌体,取1ml菌液于1.5ml 离心管,8000rpm离心3min,去上清,再取1ml 1倍PBS漂洗,去上清,吸干水分,65℃烘箱中烘约30min,至半干燥2、液氮充分研磨后,加入0.3 ml 3% CTAB提取缓冲液;3、65℃水浴45 min,取上清转到离心管中,加入40 μl 1 mg/ml蛋白酶K,15ul 10mg/ml 溶菌酶,37℃水浴30min;4、加入350 μl Tris饱和酚,摇匀,13000 r/min离心10min;5、取上清,加入等体积的氯仿/异戊醇,摇匀,13000 r/min离心10 min;6、1/10体积3M醋酸,等体积异丙醇沉淀1h7、离心,弃上清,70%酒精洗涤1-2次。
8、弃上清,晾干后用ddH2ORanase水溶解。
附效果:左边:DNA扩增16SrDNA 中间:Marker 2000bp-100bp 右边:DNA 效果图参考:1、吴发红,黄东益,黄小龙等,几种真菌DNA提取方法的比较,中国农学通报20092、scau 农学院分子遗传实验室历届研究生内部资料。
后记:参考吴等人的方法,其步骤更为复杂,要用到大离心管,小量抽提DNA不太可取,若小量抽提用他们的方法,结果是没有DNA.故此我做了两点改变:1、65℃水浴后,直接加酶,37℃温浴,时间可以不变,2、第一次氯仿:异戊醇抽提后,直接用异丙醇沉淀,酒精洗涤。
省时,且能保证DNA的量。
另外我多了溶菌酶,没做对照,也不知道有没有起作用,如果实验室没有条件,也可以不加试试。
链霉菌基因实验操作

链霉菌实验操作1录入时间:2009/8/28 16:09:31 来源:食品科技网一、培养基、抗生素、生长因子常用抗生素及其使用浓度抗生素英文名称及缩写抗性基因贮藏液浓度(mg/ml)使用终浓度(μg/ml)链霉菌大肠杆菌MM2CMYEMELA或LB氨苄青霉素氯霉素潮霉素卡那霉素壮观霉素链霉素硫链丝菌素红霉素阿泊拉霉素Ampicillin, AmpChloramphenicol, CmlHygromycin, HygKanamycin, KmSpectinomycin, SpcStreptomycin, StrThiostrepton, ThioErythomycin, EryApramycin, Amblacathygaac或aphaadAstrtsrerm Eaac(3)IV10025(无水乙醇配)5025505025(DMSO配)10050R10102?501051010R-2550502510-50R---50-2.5-5050-1002550505025不敏感2030-5*–表示无记录或不能使用,贮存液除特别说明外均用无菌水配制*此表仅供参考!!!*R表示不敏感*Km 和Am有交叉抗性,所以同时具有这两种抗性基因时应适当提高抗生素的量,并作好对照。
*Hyg易见光分解,应用锡箔纸包好。
*有些抗生素需要在低盐的环境(如DNA培养基)下筛选效率较高,如Hyg, Km, VioLB(Luria-Bertani)培养基胰蛋白胨10g,酵母抽提物5g,NaCl 5g,葡萄糖1g,蒸馏水加至1000ml,pH7.3左右(灭菌后第一次用时还要调一次)LALB中加入终浓度为1.5-2%的琼脂粉(不同的琼脂加的量不同,如青岛琼脂大概需1.5%,而华美的需要2%)。
*做蓝白斑筛选时不加葡萄糖基本培养基(MM)溶液:L-天冬酰胺0.5g,K2HPO4 0.5g,MgSO4·7H2O 0.2g,FeSO4·7H2O 0.01g,蒸馏水至1000ml将每250ml溶液分装到装有2.5g琼脂的500ml三角瓶中,灭菌,使用时每瓶加入50%葡萄糖(8磅灭菌)5ml,调pH7.0。
链霉菌总DNA提取
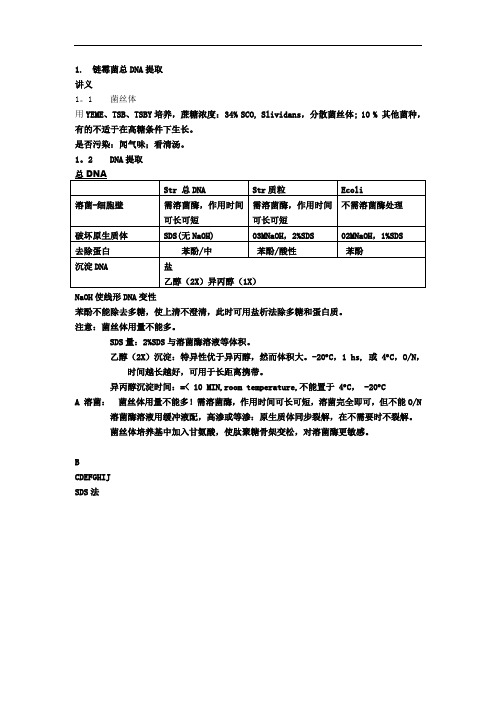
1. 链霉菌总DNA提取讲义1。
1 菌丝体用YEME、TSB、TSBY培养,蔗糖浓度:34% SCO, Slividans,分散菌丝体; 10 % 其他菌种,有的不适于在高糖条件下生长。
是否污染:闻气味;看清汤。
1。
2 DNA提取NaOH使线形DNA变性苯酚不能除去多糖,使上清不澄清,此时可用盐析法除多糖和蛋白质。
注意:菌丝体用量不能多。
SDS量:2%SDS与溶菌酶溶液等体积。
乙醇(2X)沉淀:特异性优于异丙醇,然而体积大。
-20︒C,1 hs, 或 4︒C,O/N,时间越长越好,可用于长距离携带。
异丙醇沉淀时间:=< 10 MIN,room temperature,不能置于 4︒C, -20︒CA 溶菌:菌丝体用量不能多!需溶菌酶,作用时间可长可短,溶菌完全即可,但不能O/N溶菌酶溶液用缓冲液配,高渗或等渗:原生质体同步裂解,在不需要时不裂解。
菌丝体培养基中加入甘氨酸,使肽聚糖骨架变松,对溶菌酶更敏感。
BCDEFGHIJSDS法2.9.1 链霉菌总DNA少量快速提取收集链霉菌菌丝体并用10.3%蔗糖洗涤1次,将100ul菌丝体在eppendorf离心管中用TE洗涤1次, 于菌体沉淀块中加入500ul溶菌酶溶液, 用自动移液器吸管头快速、强烈抽吸以悬浮和裂解细胞直到溶液变粘稠。
加入50ul 20% SDS,(或100ul 10% SDS)混匀, 37℃保温10分钟后, 加入0.5倍体积的中性苯酚/氯仿, 混匀后离心, 向上清液加入0.1倍体积NaAC(pH4.8)和等体积异丙醇, 颠倒混匀, 将白色絮状沉淀挑出到70%乙醇中洗涤, 离心, DNA沉淀块再分别用70%乙醇和无水乙醇洗涤后溶解在适量TE缓冲液中。
2.9.2 大片段链霉菌总DNA提取(用于构建基因文库)适量的菌丝体(50ml液体培养物), 溶于10ml含2mg/ml溶菌酶的LRTE溶液(2mg/ml溶菌酶, 25mM Tris-HCl pH8.0), 100mM EDTA (pH8.07, RNase 50ug/ml)中, 37℃溶菌至溶液清澈透亮。
一种高效可直接用于PCR扩增的不吸水链霉菌基因组DNA的提取方法

生物技术通报BIOTECHNOLOGYBULLETIN・研究报告・2008年第4期收稿日期:2008-02-26作者简介:吴红艳(1967-),女,副研究员,研究方向:微生物及其分子生物学研究不吸水链霉菌是由辽宁省微生物科学研究院在广西梧州地区土样中分离得到一株不吸水链霉菌(Streptomycesahygroscopicus),其经发酵产生的次生代谢产物为广谱抗真菌抗生素(命名为梧宁霉素),对多种植物病原菌具有较强的抑制和杀死作用,而且其毒性很低,具有广阔开发应用前景。
但其一直存在着产量低、效价低、保存期短等问题,阻碍其广泛的推广应用,这就需要找到一种改善的方法。
随着分子生物学技术的不断提高,克隆不吸水链霉菌次级代谢过程中的激活因子和限速酶基因,增加其在菌种次级代谢中的表达,要对目的基因进行表达,首要的任务就是基因组DNA或RNA的提取,然后进行PCR扩增、PCR产物经回收、纯化后与载体连接、转入宿主细胞中进行诱导表达以及表达产物相关指标的检测等,因而其基因组DNA或RNA的获得是前提条件。
下面对于不吸水链霉菌基因组DNA的提取方法进行了研究探讨。
1材料与方法1.1材料1.1.1菌种不吸水链霉菌菌种由辽宁省微生物科学研究院菌种中心提供。
1.1.2试剂TE缓冲液、裂解液、蛋白酶K、无水乙醇、苯酚、氯仿、异戊醇、异丙醇、亚精胺、pvp、Taq酶、dNTP等。
1.1.3仪器高速冷冻离心机、水浴锅、电泳仪、PCR仪等。
1.2方法1.2.1不吸水链霉菌基因组DNA提取。
一种高效可直接用于PCR扩增的不吸水链霉菌基因组DNA的提取方法吴红艳陈飞桓明辉关艳丽郭玲玲(辽宁省微生物科学研究院,沈阳122000)摘要:通过对提取不吸水链霉菌基因组DNA的冻融法、微波法和溶菌酶破壁法等几种方法进行对比、分析,得出结论:溶菌酶破壁法所提取的不吸水链霉菌基因组DNA可直接用于PCR扩增。
这为今后不吸水链霉菌相关基因的实验研究提供了可靠的理论依据。
一种高效提取真菌总DNA的方法

M ycosystema菌 物 学 报 15 March 2009, 28(2): 299-302jwxt@ISSN1672-6472 CN11-5180Q©2009 Institute of Microbiology, CAS, all rights reserved.基金项目:国家自然科学基金(No. 30271085);黑龙江省自然科学基金(No. 220-413504);哈尔滨市青年科学基金项目(No. 2002AFQXJ006)*Corresponding author. E-mail: peikequan@收稿日期: 2008-03-26, 接受日期: 2008-04-23一种高效提取真菌总DNA 的方法孙立夫1,2 张艳华1 裴克全2*1绍兴文理学院生命科学学院 绍兴 3120002中国科学院植物研究所植被与环境变化重点实验室 北京 100093A rapid extraction of genomic DNA from fungiSUN Li-Fu 1, 2 ZHANG Yan-Hua 1 PEI Ke-Quan 2*1Life Science Faculty, Shaoxing College of Arts & Sciences, Shaoxing 312000, China2Key Laboratory of Vegetation and Environmental Change, Institute of Botany, The Chinese Academy of Sciences, Beijing 100093, China在真菌的分子生物学研究中,快速高效地提取质量优良的DNA 有着重要意义。
目前使用的真菌DNA 提取方法步骤大体相似,主要差别在于采用何种方法来打破细胞壁这一关键环节。
常用的破壁方法主要有冷冻干燥研磨法、玻璃珠机械破壁法、酶解法和氯化苄法等(吴志红 2001)。
细菌总DNA提取原理与方法

2. 3. 4.
加入1 ml 20%SDS,65℃水浴过夜 加入1/3倍体积饱和Nacl,剧烈振荡,12000 r/min 离心10min 取上清,用等体积苯酚:氯仿:异戊醇各抽提1次,水相中加入 等体积TE,再加入0.6倍体积的异丙醇室温沉淀DNA 2h
5.
12000 r/min 离心10min收集DNA沉淀,用200μlTE溶解
常用的电泳缓冲液
4℃ 保存备用
常用的6X载样缓冲液
Ⅰ 0.25%溴酚蓝 ,0.25%二甲苯青 ,40%蔗糖水溶液 Ⅱ 0.25%溴酚蓝 ,0.25%二甲苯青 ,30%甘油水溶液
2.
[实验步骤和过程] (一)细菌总 DNAD 提取 革兰氏阳性菌DNA提取方法(提取基因组DNA) 1. 3-6ml 细菌过夜培养液, 5000rpm离心10分钟, 弃上清 2. 加0.5ml TE悬浮沉淀,并加75μl 溶菌酶(20mg/ml) 溶液,40°C保温30分钟 3. 加50μl, 10% SDS, 5μl 蛋白酶 K (20mg/ml),混 匀, 37℃保温30分钟 4. 加0.75ml, 5mol/L NaCl, 混匀 5. 用等体积酚:氯仿:异戊醇(25:24:1)抽提, 5000rpm离 心10分钟, 将上清液移至干净离心管
方法四:
1. 取20μl TE、 20μl 饱和酚与无菌小离心管中 2. 取已经培养好的菌一环到上述离心管中 3. 震荡离心管30秒 4. 离心, 8000rpm, 10min 5. 取上清 3-5 μl 进行琼脂糖凝胶电泳检测
(二)DNA的琼脂糖凝胶电泳 琼脂糖是一种天然聚合长链状分子,可以形成 具有刚性的滤孔,凝胶孔径的大小决定于琼脂 糖的浓度 DNA分子在碱性缓冲液中带负电荷,在外加电 场作用下向正极泳动 DNA分子在琼脂糖凝胶中泳动时,有电荷效 应与分子筛效应。不同DNA的分子量大小及构 型不同,电泳时的泳动率就不同,从而分出不 同的区带
链霉菌基因组DNA提取
链霉菌基因组DNA提取
1.刮取孢子接种于TSB中,培养菌体4-5d,吸取2ml的菌液于
2.0ml离心管中,3000rpm
离心15min收集菌体;
2.加入ddH2O洗涤菌体两次;
3.用500μl溶菌酶溶液(加RNaseA)重新悬浮菌丝体,于37℃温育约50min至细胞成为
半透明状(中途取出涡旋混匀几次);
4.加入500μl 2%SDS,混合振荡约lmin直到溶液的粘度显著下降,打开盖子,于55℃温育
30min;
5.冷却至室温,然后加入1/10体积3M醋酸钠和200μl氯仿/异戊醇(24:1),涡旋振荡约
1min后,4℃12000rpm离心5min;
6.小心地吸取上清液,弃去白色中间层。
(不要吸到中间白色物质),用氯仿/异戊醇(24:1)
重复二次抽提直至看不见(或非常少)中间层为止;
7.取上清,加入预冷的2倍体积的无水乙醇,反复颠倒,-20℃放置30min.置絮状DNA沉
淀出现;
8.离心倾去上清液,DNA沉淀块分别经70%乙醇洗涤,用枪头吸去液体,晾干数分钟;
9.后溶解在适量的ddH2O中,用分光光度计测浓度及纯度后,-20℃保存。
霉菌提取基因组DNA方法
CTAB法提取真菌基因组DNA1. 取100mg菌体至预先装有300mg石英沙的1.5ml离心管中;2.在离心管中加入500ul 2X CTAB裂解液(2% CTAB W/V,100mmol/L Tris·Cl, pH8.0, 20mmol/L EDTA, 1.4mol/L NaCl);3.用专用塑料槌充分研磨得到均一的浆状物;4. 60℃水浴1 小时(10分钟翻转离心管一次);5. 加入2/3体积的苯酚/氯仿/异戊醇溶液(25:24:1), 颠倒混匀(需带手套, 防止损伤皮肤),室温下静置5-10分钟, 使水相和有机相分层(必要时可重新混匀)。
6. 室温下13000rpm离心30分钟;7. 仔细移取上清液至另一1.5ml离心管,重复5-7步骤3-5次,直到两个界面没有沉淀;8.小心加入2倍体积的100%的冷乙醇(-20℃预冷过夜);9.-20℃放置3个小时或者过夜;10.4度,13000rpm离心30分钟;11.小心倾去上清,将离心管压在干净吸水纸上吸干;12.沿离心管内壁缓慢加入200ul 70%的冷乙醇(4度),尽量把壁上的沉淀洗入管底;13.11000rpm,4℃离心20min;倾去上清,晾干;14. 加入50-100ul TE(10mmol/L Tris·Cl, pH8.0, 1mmol/L EDTA,300µg Rnase)到离心管中;15. 37℃水浴30min, 除去RNA;16. 重复5-13步骤,加入100ul TE溶解, -20℃贮存。
17. 取2μl DNA样品在0.7% Agarose胶上电泳, 检测DNA的分子大小。
同时取10μl 稀释20倍, 测定OD260/OD280, 检测DNA含量及质量。
链霉菌RNA提取
5.5.10 转录谱的测定5.5.10.1 链霉菌总RNA的提取①液体培养基中生长的菌体经离心收集并用吸水纸洗干(固体培养时,应在培养基表面铺上玻璃纸,刮取生长在玻璃纸表面的菌体),迅速放入用液氮预冷的研钵中,研磨至粉末状;②取50-100 mg菌体放入预冷的无RNase污染的离心管中(15磅灭菌1小时),迅速加入1 ml Trizol试剂,用微量移液器吹匀并在振荡器上振荡2分钟,室温放置5分钟后12,000 g,4°C离心10 min;③将上清转移至一新的离心管中,加入200 μl氯仿,充分混匀,室温放3分钟后于12,000 g,4°C离心15 min;④将上清转移至一新的离心管中,再加入200 μl氯仿抽提一次,12,000 g,4°C离心10 min;⑤将上清转移至一新的离心管中,加入500 μl异丙醇,-20°C或-70°C沉淀30 min,12,000 g,4°C离心10 min;⑥弃上清,加入1 ml 70%乙醇洗两次,室温干燥后加入适当体积的DEPC处理过的H2O溶解,-70°C冻存,或用甲酰胺溶解RNA沉淀贮存于-20°C。
5.5.10.2 DNase处理RNA样品RNA样品用RQ-RNase free DNase(50 μl体系加1 μl 酶)处理1 h,加水至500 μl,酚、氯仿抽提后,加1/10体积的3 M醋酸钠(pH 5.0),2倍体积的无水乙醇沉淀,最后溶解于适量的水中。
PCR是否有DNA污染。
5.5.10.3 RNA浓度和纯度的检测①利用紫外分光光度计测定所提取RNA在260和280 nm的光吸收值,确定RNA的纯度和浓度。
纯净RNA样品的OD260/OD280比值应介于1.9和2.05之间,比值小于1.8表明蛋白污染,大于2.1可能是降解严重。
根据OD260为1相当于40 μg/ml RNA来计算浓度。
两种pHZ1358衍生物提高链霉菌基因敲除的效率
推进链霉素基因敲除效率两种pHZ1358衍生物【摘要】链霉菌质粒pIJ101中sti基因的删除使其pHZ1358衍生物成为基因破坏和代替的有效载体。
现在看来,pHZ1358被衍生的质粒pJTU1278进一步优化了,这种衍生质粒多为一个携带多个克隆位点和LacZ基因筛选标记的盒子,方便了在大肠杆菌中质粒的产生。
此外,在pJTU1278的oriT区域也已被删除,产生一个载体(pJTU1289),其可用于PCR定位的明确。
这些载体的有效使用,证明了该基因删除与阿维链霉菌中阿维菌素的生物合成有关。
【关键字】穿梭载体pHZ1358, pJTU1278, pJTU1289,共轭聚合酶链反应定位链霉菌,土栖革兰氏阳性菌,因他们有能力生产出多种生物活性化合物,故在生物技术研究领域具有重要的意义。
链霉菌也不同于一般的原核生物,因为它们得经历一个复杂的形态分化周期,在其生命周期的不同阶段形成孢子,营养基质菌丝和气生菌丝。
尽管为了不同的研究目的而不断对这一方面开发新技术,但是基因缺失或基因崩解仍然是最有效的也是不可缺少的手段之一。
为了对链霉菌类基因进行干扰和替代,pHZ1358已发展为一个有效率的pIJ101衍生载体,通过去除一个含有导致宿主体内积累的单链质粒的sti(强效的不相容轨迹)的DNA区域。
36 - bp直接重复的进一步删除,使复制的质粒可在大肠杆菌里作一稳定的穿梭载体在这里,我们报告两个pHZ1358衍生物,pJTU1278和pJTU1289,其具有复合多样的克隆位点和一个LacZ基因筛选标记(为大肠杆菌合成便捷),而后者已删除oriT,允许它是专门用于PCR的靶向点。
对pJTU1289的使用情况也通过阿维菌素生物合成基因的删除得以证明。
材料和方法本研究中运用了菌株和质粒DH10B(GIBCO - BRL)和ET12567(pUZ8002)分别被用来作为大肠杆菌的克隆和共轭。
而阿维链霉菌NRRL 8165则用于基因删除。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1. 链霉菌总DNA提取
讲义
1。
1 菌丝体
用YEME、TSB、TSBY培养,蔗糖浓度:34% SCO, Slividans,分散菌丝体; 10 % 其他菌种,有的不适于在高糖条件下生长。
是否污染:闻气味;看清汤。
1。
2 DNA提取
NaOH使线形DNA变性
苯酚不能除去多糖,使上清不澄清,此时可用盐析法除多糖和蛋白质。
注意:菌丝体用量不能多。
SDS量:2%SDS与溶菌酶溶液等体积。
乙醇(2X)沉淀:特异性优于异丙醇,然而体积大。
-20︒C,1 hs, 或 4︒C,O/N,时间越长越好,可用于长距离携带。
异丙醇沉淀时间:=< 10 MIN,room temperature,不能置于 4︒C, -20︒C
A 溶菌:菌丝体用量不能多!需溶菌酶,作用时间可长可短,溶菌完全即可,但不能O/N
溶菌酶溶液用缓冲液配,高渗或等渗:原生质体同步裂解,在不需要时不裂解。
菌丝体培养基中加入甘氨酸,使肽聚糖骨架变松,对溶菌酶更敏感。
B
CDEFGHIJ
SDS法
2.9.1 链霉菌总DNA少量快速提取
收集链霉菌菌丝体并用10.3%蔗糖洗涤1次,将100ul菌丝体在eppendorf离心管中用TE洗涤1次, 于菌体沉淀块中加入500ul溶菌酶溶液, 用自动移液器吸管头快速、强烈抽吸以悬浮和裂解细胞直到溶液变粘稠。
加入50ul 20% SDS,(或100ul 10% SDS)混匀, 37℃保温10分钟后, 加入0.5倍体积的中性苯酚/氯仿, 混匀后离心, 向上清液加入0.1倍体积NaAC(pH4.8)和等体积异丙醇, 颠倒混匀, 将白色絮状沉淀挑出到70%乙醇中洗涤, 离心, DNA沉淀块再分别用70%乙醇和无水乙醇洗涤后溶解在适量TE缓冲液中。
2.9.2 大片段链霉菌总DNA提取(用于构建基因文库)
适量的菌丝体(50ml液体培养物), 溶于10ml含2mg/ml溶菌酶的LRTE溶液(2mg/ml溶菌酶, 25mM Tris-HCl pH8.0), 100mM EDTA (pH8.07, RNase 50ug/ml)中, 37℃溶菌至溶液清澈透亮。
加入蛋白酶E至终浓度为0.2mg/ml, 小心轻轻混匀, 30℃, 30分钟, 加入20%SDS 至终浓度为1%, 立即轻轻倾斜混匀, 37℃水浴2小时。
冷却至室温, 加入等体积中性苯酚, 轻轻混匀(约需10分钟), 使液体均一相形成乳浊液, 5000g离心15分钟, 用大口径移液管转移上清液, 根据中间蛋白质的多少, 重复使用中性苯酚抽提。
经中性苯酚抽提了的上清液, 再用氯仿抽提两次。
最后向上清液中加入0.4倍体积的7.5M NH4AC和两倍体积的无水乙醇, 小心转动离心管以充分混匀, DNA立即形成白色絮状沉淀。
小心挑出絮状沉淀团到5ml 70%乙醇中洗涤两次, 再用无水乙醇洗涤1次, 自然风干(勿使沉淀完全干燥, 否则极其难溶), 加入适量TE缓冲液, 4℃过夜溶解DNA。
经脉冲电场凝胶电泳检测发现用这种方法提取的总DNA片段在90-160kb之间。
DNA溶液中蛋白质及RNA的去除
一般是用Tris饱和的苯酚或苯酚/氯仿抽提DNA溶液以除去溶液中的蛋白质。
向DNA 溶液中加入等体积的Tris饱和的苯酚或苯酚/氯仿(pH7.8-8.0), 若DNA分子小于10kb, 可采用剧烈振荡的方式充分混合有机相与水相, 若DNA分子在10-30kb之间, 可通过轻轻摇动进行混合, 若DNA分子大于30kb, 则需要轻轻转动和颠倒进行混合, 以免DNA被剪切。
离心分离两相, 取上层的水相(DNA分子大于30kb时, 应使用大口径吸管), 重复抽提数次直至有机相与水相之间看不到白色物为止。
最后用等体积氯仿抽提两次以除去可能残留在溶液中的苯酚。
对于DNA样品中存留的的RNA, 可以通过加入0.1倍体积的8M LiCl, 混合后置冰浴中30分钟, 15000g离心10分钟, 取上清液而弃去沉淀物(大分子RNA), 再向上清液中加入RNase至终浓度为20ug/ml, 37℃, 30分钟, 即可沉淀DNA。
有时在提取质粒和总DNA时, 向粗提液中加入RNase到终浓度为10ug/ml, 37℃水浴30分钟后, 用上述方法去除蛋白质。