成人多囊肾PKD1和PKD2
【案例分享】一例多囊肾的基因检测

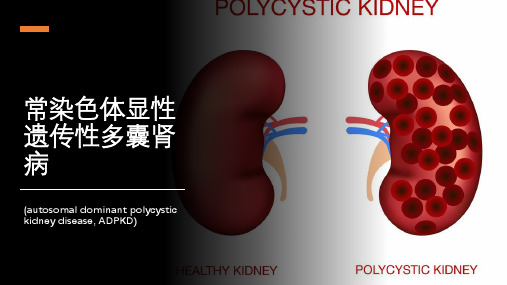
【案例分享】⼀例多囊肾的基因检测概述多囊肾病(PKD)是⼈类常见的单基因遗传病之⼀,按遗传⽅式可分为常染⾊体显性多囊肾病(ADPKD)和常染⾊体隐性多囊肾病(ARPKD)。
常染⾊体隐性多囊肾,是幼⼉型多囊肾,发病于婴⼉期,临床较罕见,发病率1/10000-1/40000;常染⾊体显性多囊肾,是成⼈型多囊肾,常于青中年时期被发现,也可在任何年龄发病,为多囊肾的常见类型,发病率为1/400-1/1000。
多囊肾病(PKD)的特征是肾中液体囊肿。
这是慢性肾功能不全和终末期肾病的第四⼤常见原因。
除累及肾脏外,常伴肾外囊肿,如肝囊肿、胰腺囊肿、脾囊肿等。
常染⾊体显性多囊肾病(ADPKD)是最常见的危及⽣命的遗传性疾病,其特征在于囊肿形成及肾脏和其他器官的肿⼤。
在ADPKD中有两种已知的基因突变:PKD1基因和PKD2基因。
PKD1是编码多囊蛋⽩1的⼤⽽复杂的基因,PKD2较⼩,编码多囊蛋⽩2。
ADPKD与其他囊性肾病的鉴别诊断取决于患者的年龄,家族史和相关表现。
在没有ADPKD家族史的成⼈患者中,医⽣应排除常染⾊体隐性多囊肾病(ARPKD)。
幼⼉在没有ADPKD家族史的情况下,重要的是区分ARPKD。
常染⾊体隐性多囊肾病(ARPKD)通常⼜被成为婴⼉性多囊肾病,属于肝肾纤维囊性疾病,典型者在婴⼉期发病,同时也是⼉科终末期肾脏病的重要原因之⼀,⼈群携带者为1:70。
30.5-50%的患病胎⼉因⽺⽔过少导致肺发育不全⽽在围产期死亡,此期主要为肾损害。
影像学检查主要表现为双肾集合管的⾮梗阻性梭形膨⼤,回声超强,呈陶⼟样“Potter”⽺⽔过少症。
如果度过新⽣⼉期,⼀般患⼉会进展⾄终末期肾病。
症状体征ADPKD主要表现为双侧肾囊肿且逐渐发展,肾脏体积进⾏性增⼤,肾⼩球滤过率逐步降低,常出现⾼⾎压(60%)、腹痛(61%)、⾎尿(15%)、蛋⽩尿(11%)、囊肿或尿路感染、肾结⽯(10%-35%)等并发症。
此外,ADPKD属多系统性疾病,35岁以上患者中超过90%合并肝囊肿、颅内动脉瘤、胰腺囊肿、结肠憩室或⼼脏瓣膜异常;其中肝囊肿是最常见合并症(83%-94%)。
PKD1基因新突变导致常染色体显性多囊肾病

4.蒋春波.复发性尿路感 染免 疫机 制研究 意义 及进 展.中 国中
— 851.
西医结合 肾病杂 志 ,2010,11(8):735—737.
(收稿 :2017—03—28 修 回 :2017—05—28)
5.郑筱萸.中药新药I临床研究指导原则 (试 行).北京 :中国医药
PKD1基 因新 突变 导致 常 染 色体 显 性 多囊 肾病
机制提供有力证据 ,并 为防治慢性复杂性疾病 的临床治疗 提供 尿路感染 的效果观察 .当代 医药论丛 ,2015,13(24):32—34.
指 导 。
7.张众.栝楼瞿麦 丸加减治 疗糖 尿病神 经源性 膀胱疗 效 观察.
参 考 文 献
实 用 中医 药 杂 志 ,2011,27(7):452—453. 8.刘晨 ,张凌珲 ,杨 柳 ,等.瞿 麦药学研 究概 况.安 徽农业科 学 ,
—
[关键词 ] 常染 色体显性 多囊肾病 PKD1 基 因突变 新 一代 测序技 术
常染色体显性 多囊 肾病 (autosomal dominant polycystic kid— hey disease,ADPKD)是最 常见 的遗传性肾病 ,发病率为 1/400~ 1/1 000。该病 主要表 现 为双侧 肾脏 形 成多 个液性 囊 泡 ,囊泡
张慧峰① 于为 民① 郭永艳 ② 吕 艳 ① 尚 雁 ① 薛晋 杰③
[摘 要 ] 目的 :确定 1例常染 色体显性 多囊肾病 (ADPKD)患者致病基 因的 突变 ,以明确诊 断和指导 患者 生育。方 法 : 采 用高通 量测序 (next generation sequencing,NGS)技 术对患者 3个 多囊肾致病基 因进行检 测 ,并通过 Sanger测序 进行验 证。 结果 :患者 PKD1基 因存在插入 突变 C.11853—11854 insA,从 而使 其编码 蛋 白丧失 功能。结论 :PKD1基 因新 的致病 突 变 C. 11853 11854 insA的发现 丰富 了 ADPKD的发 生机 制 ,临床上 可通过产前诊 断或植入 前遗传 学诊 断预 防患者再生育 患儿 。
《多囊肾病》PPT课件

两项主要+一项次要
第一项主要+三项以上次要
医学PPT
17
诊断
3、影像学检查: Ravine等于1994年提出了一下B超诊断标准:
有家族遗传史的30岁以下患者,单侧或双侧肾脏 有2个囊肿,30~59岁患者双侧肾脏至少各4个囊 肿;如果同时伴有其他肾外表现,如肝囊肿等, 诊断标准可适当放宽。此诊断标准敏感性97%, 特异性90%。如无家族遗传史,每侧肾脏有10个 以上囊肿,并排除其他肾囊肿性疾病方可诊断
多囊肾病
(polycystic kidney disease,PKD)
医学PPT
1
• 肾囊肿:包曼囊~集合管间的局部扩张→与 原来的肾小管脱离→独立、充满液体的囊肿
• 囊性肾脏病:肾脏出现单个或多个囊肿的一 组疾病,分遗传性和非遗传性
医学PPT
2
医学PPT
3
多囊肾的分型
目前已知有3种基因的突变导致 了ADPKD
其中2种最为常见: 为PKD1 (约 占85% )和PKD2 (约占15% )
第3种是PKD3
医学PPT
4
多囊肾的分型
• 常染色体显性遗传型,( ADPKD)此型一般到成年 才出现症状,又称成人型 多囊肾,实际上可发生于 任何年龄,故“成人型”并 不准确,已废用
• 常染色体隐性遗传型,又 称婴儿型多囊肾. 一般在 婴儿即表现明显
医学PPT
13
发病机制2、螺旋区-螺旋区相互作 用假说
医学PPT
14
发病机制3、纤毛致病学说(中心地位)
纤毛无运动功能,必须依赖一个非常重要 的生理过程———鞭毛内运输(IFT) 来完成, 驱动蛋白- 和细胞浆动力蛋白- 1B 作为2种 分子动力蛋白, 分别参与其中的顺向转运和 逆向转运过程,纤毛通过多囊蛋白复合体感 受尿流率调控肾小管腔直径和分化状态, 基 因突变引发了纤毛感受尿流率的功能异常, 可通过下游Ca2 +信号传导影响靶基因调控 ,进而导致细胞功能改变和囊肿生成
关于胡桃夹综合征与多囊肾

胡桃夹综合征(nutcracker phenomenon)即(左肾静脉压迫综合征),又称胡桃夹现象,好发于青春期至40岁左右的男性,儿童发病分布在4~7岁,多发年龄见于13~16岁。
人体的血管像四通八达的道路一样,是有一定走向的。
左肾静脉行走在腹主动脉和肠系膜上动脉之间,这两条动脉构成40~60度的夹角,左肾静脉刚好通过此夹角。
从解剖上看,右肾静脉径直注入下腔静脉,行程短而直。
而左肾静脉则需穿过腹主动脉和肠系膜上动脉之间的夹角,跨越腹主动脉前方始能注入下腔静脉,因此左肾静脉远较右肾静脉长。
正常时,肠系膜上动脉与腹主动脉之间的夹角被肠系膜、脂肪、淋巴结和腹膜等所充塞,使左肾静脉不致受到压挤。
当青春期发育较快、身高迅速增长、脊柱过度伸展、体形急剧变化或肾下垂等情况下,左肾静脉在这个夹角中的日子就不好过了,会受到挤压,引起血流变化和相应的临床症状。
症状:胡桃夹现象的主要症状是血尿和蛋白尿,其中无症状肉眼血尿更易发现。
血尿的原因是左肾静脉受压致肾静脉高压,左肾静脉扩张所引流的输尿管周围静脉与生殖静脉淤血、与肾集合系统发生异常交通,或部分静脉壁变薄破裂,引起非肾小球性血尿,还会发生睾丸静脉和卵巢静脉淤血而出现肋腹痛,并于立位或行走时加重。
另外男性还能发生精索静脉曲张。
此外有蛋白尿,不规则月经出血,高血压等。
此病的诊断标准为:一侧肾出血;尿红细胞形态为非肾小球性;尿中钙排泄量正常;膀胱镜检查为左侧输尿管口喷血或血性尿;腹部彩超或CT检查可见左肾静脉扩张等。
超声检查:超声对胡桃夹综合征的诊断有着明显的优势,超声检查时可清晰显示腹主动脉、肠系膜上动脉及左肾静脉的解剖情况,在不同横断面均可找到左肾静脉扩张近段的最大内径,测值准确,同时可观察并测量肠系膜上动脉与腹主动脉夹角变化。
彩超血流速度提供更准确的血流动力学变化,有助于此病诊断。
超声检查还能除外先天性畸形、外伤、肿瘤、结石、感染性疾病及血管异常等造成的血尿。
多囊肾多囊肾病(polycystic kidney disease,PKD)是一种常见的遗传性肾脏病,主要表现为双侧肾脏出现多个大小不一的囊肿,囊肿进行性增大,最终破坏肾脏结构和功能,导致终末期肾功能衰竭。
常染色体显性多囊肾

常染色体显性多囊肾(Autosomal dominant polycystic kidney disease,ADPKD)又称为成人型多囊肾,是一种遗传性全身性疾病,主要影响肾脏,但也可能会影响其他器官,如肝脏、胰腺、脑动脉血管等。
患有这种疾病的人大约有一半将会发展为终末期肾脏疾病,需要进行透析或肾移植。
患者进展为终末期肾病通常发生在40-60岁之间[1]。
常染色体显性多囊肾病在全球范围都有发生,发病率大约为1/400人-1/1000人[2][3]。
现在认为,常染色体显性多囊肾和两个基因缺陷有关。
85%的患者是由位于16号染色体的基因PKD1(TRPP1)发生突变所致,15%的患者是由PKD2(TRPP2)突变所致[1]。
常染色体显性多囊肾需要和常染色体隐性多囊肾进行辨别。
常染色体隐性多囊肾也导致肾脏和肝脏的囊肿,但通常只发生在童年,发病率约为1/20000人[4],病因和预后都和常染色体显性多囊不同。
目录 [隐藏]1 病理生理学2 诊断3 治疗4 预后5 参考资料6 外部链接病理生理学 [编辑]近期应用实验模式生物如线虫(秀丽隐杆线虫)和家鼠的纤毛和鞭毛进行基本细胞生物学研究使人类发生常染色体显性多囊肾的原因变得清楚。
生化学家James Calvet写到:“这些发现证明了遗传学和动物模型的能力和重要性。
如果没有这些遗传学研究的指引,有谁会想到一条小小的纤毛会成为研究多囊肾病的基础?”[5]纤毛在肾脏发育中发挥重要作用,纤毛结构功能异常直接导致肾囊肿性疾病的发生。
所有的纤毛和鞭毛的装配和维持都需要依赖鞭毛内运输这个非常重要的生理过程来完成。
鞭毛内运输是蛋白质插入纤毛和鞭毛膜特定位点必不可少的一项细胞功能。
这些插入的膜蛋白可以启动环境反馈和细胞内信号转导通道。
它们在肾小管上皮细胞的纤毛中发挥特殊作用,通过上述鞭毛内运输机制定位于肾小管上皮细胞的纤毛,被认为是正常肾细胞的发育和发挥功能的关键。
纤毛上皮细胞排列于尿收集管的内腔,感觉尿流率的变化。
常染色体显性遗传性多囊肾病
流行病学ห้องสมุดไป่ตู้
•
所有种族都可发生ADPKD,报道的患病率为1:1000。在美国每年
开始透析的患者中,大约有5%的肾病由ADPKD引起。
•
ADPKD主要由两种基因突变引起:16号染色体上的PKD1(编码多
囊蛋白-1)或4号染色体上的PKD2(编码多囊蛋白-2)。大多数患者可维持
正常肾功能到30岁左右。一旦GFR开始下降,其每年可平均降低4.4-
➢ 多囊肝 多数不需治疗。肝脏明显增大可引起腹胀、呼吸困难、胃食管反流、门静脉高压等。可根据病情选 择肝囊肿穿刺硬化、去顶减压术、肝部分切除术或肝移植术
➢ 颅内动脉瘤 对于有动脉瘤和蛛网膜下腔出血家族史的病人,推荐MRI血管造影检查确诊。直径大于10mm的动 脉瘤应采取介入或手术治疗
治疗
肾脏替代治疗 ➢ 包括血液透析、腹膜透析和肾移植。目前认为ADPKD病人腹膜透析与血液透析的并发症和 长期存活率无明显差异。移植后肾存活率、并发症与其他肾移植人群相似
治疗
对症治疗 ➢ 疼痛
急性疼痛针对病因进行治疗。慢性疼痛,程度轻者或一过性疼痛卧床休息并观察,如疼痛持续或较 重按止痛阶梯序贯药物治疗,仍不能缓解可考虑囊肿穿刺硬化、囊肿去顶减压术及多囊肾切除术
➢ 出血 多为囊肿出血所致,呈自限性,轻者绝对卧床休息、止痛、多饮水。出血量大、保守疗法效果差可 行选择性血管栓塞或出血侧肾脏切除
泌尿道和囊肿感染,引起ADPKD病人发热的首要病因,主要表现为膀胱炎、肾盂肾炎、囊肿感 染和肾周脓肿,逆行性感染为主要途径
临床表现
肾外表现 ➢ 囊性病变
可累及肝脏、胰腺、脾脏、卵巢及蛛网膜等器官。其中肝囊肿最常见,大多数病人无症状,少数 可表现为疼痛、囊肿感染和出血
多囊肾

发病机制
正常成人单个肾脏的重量约为150g。在无症状的成年多囊肾患 者,单个多囊肾的重量平均为256g;在有症状的成年多囊肾患 者,单个多囊肾的重量平均为465g。在成年型多囊肾,肾脏常 见弥漫性囊肿,不论肾皮质和髓质均布满大小不等的囊肿,形 似一串葡萄。囊壁上皮细胞有局限性增生,形成息肉样,细胞 外基质异常增生,近端小管膨出形成的囊肿,囊液成分似血浆; 远端小管形成的囊肿,囊液中钠、氯含量较低,而尿素和肌酐 浓度较高。在有症状的多囊肾患者,随着年龄的增长,囊肿数 目增多,囊腔增大,直径达2~3cm,后期肾脏长径可达20~ 30cm,全肾几乎被囊肿所占据。
发病机制
本病发病机制未明。Hilde-brand等认为多囊肾可能是由 Bowman囊扩张而来,也可能是由肾曲小管扩张而成,系由后 肾胚芽发育而成的肾小球、肾曲小管与Wolffian管发育而成的集 合管之间的沟通受到障碍的缘故;Bialestack提出,有些囊肿则 是异常增大的肾单位,称为巨肾单位。亦有认为有些囊肿具有 排泄功能;Bricker等对囊肿中的液体进行化学分析,证明其所 含成分与尿液相近;Norris等认为是由于很多临时性的后肾单位 不能正常萎缩,一部分肾单位发生局部缩窄及分节,因而形成 大小不同的囊肿;Hepler等则认为是肾脏的血液循环分布不正 常,造成肾实质退变的结果;Hidd-brant认为分泌部(来源于肾 组织的肾曲小管及部分肾小球)与排泄部(来源于输尿管芽的集 合管、肾盂等)在发育时期彼此失去联系,分泌部成为盲端,其 分泌物无从排出,故形成多数囊肿;另有人认为是机械性因素 如胎儿时期局部炎症引起排泄管纤维阻塞,或由于管型及不溶 解性钙盐阻塞,使尿液不畅引起肾小管扩大的结果。
多囊肾
大头医生
编辑整理
英文名称
常染色体显性遗传多囊肾研究进展

常染色体显性遗传多囊肾研究进展摘要常染色体显性遗传多囊肾(ADPKD)是一种发病率高、预后差的疾病,它在发病机制、治疗等方面有很多进展。
关键词常染色体显性遗传多囊肾;瞬时受体势;Max作用因子1;雷帕霉素靶蛋白常染色体显性遗传多囊肾(autosomal dominant polycystic kidney disease,ADPKD)是一种常见的遗传性肾病,是导致肾衰竭的重要疾病。
现在已经发现3个基因(PKD1、PKD2、PKD3)与此病有关,其中PKD1定位于染色体16p13.3,其突变而导致ADPKD约占85%,PKD2定位于染色体4q21-23,其突变约占15%。
PKD3突变仅在几个家族中发现,目前尚未定位[1]。
ADPKD病变以双肾多发性进行性充液囊泡为主要特征。
囊泡损伤肾组织,引起肾功能改变,出现血尿、蛋白尿等临床症状,最终导致肾衰竭。
ADPKD除累及肾脏外,还可引起肝脏囊肿、胰腺囊肿、心脏瓣膜病、结肠憩室和颅内动脉瘤等肾外病变[2],给患者、家属及社会带来沉重负担。
故揭示其发病机理,研究新的治疗方法有很重要的意义。
本文对这些进展作以综述。
1 ADPKD的发病机制1. 1 多囊蛋白PC PKD1基因的蛋白产物被称为多囊蛋白-1(polycystin-1,PC1),又叫做TRPP1,多囊蛋白-1是一种跨膜蛋白,分布广泛,可以与多种蛋白(如PC2)、糖、脂类结合并发生交互作用,从而发挥功能。
PC1还与Wnt信号途径、JAK-STAT途径、转录因子AP-1和G蛋白偶联等信号途径有关。
PKD2基因的蛋白产物被称为多囊蛋白-2(polycystin-2,PC2),又叫做TRPP2,是TRP家族的一员,为非选择性钙离子通道。
它不同于PKD1,在人类基因组中是单拷贝,并不含多嘧啶区,但它的第一个外显子富含GC,从而使得此处易于突变[3]。
瞬时受体势(transient receptor potential,TRP)通道虽然最初发现于感受器,主要参与神经传导,但随着研究的深入,近年来人们发现该通道在肾脏病的发生、发展中也发挥了作用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
ADPKD)和常染色体隐性遗传多囊肾 病(autosomal recessive polycystic kidney disease, ARPKD)。
概述
ADPKD既往被称为成人型多囊肾病,是最常见的遗传性肾脏病之一。 特点:任何年龄均可发病,多发于30-50岁;
血尿: 30%-50%的患者出现肉眼血尿或镜下血尿,蛋白 尿一般轻度并呈持续性。
高血压: 60%的成年患者在病程中出现高血压,与肾脏 增大、囊肿扩大的程度相关,且对肾损害的进展起促进 作用。
肾功能损害:常见肾脏浓缩功能减退,表现为夜尿增多, 尿比重降低。随着年龄增长和病情发展,一半以上患者 出现慢性肾衰竭。
鉴别诊断
需要鉴别的非遗传性囊性肾病包括:
➢ 1.多发性单纯性肾囊肿:常在体检时发现,无临床症状。 无家族史,肾脏大小正常,一般累及单侧肾脏,也可累及 双侧肾脏,典型的单纯性肾囊肿分布于肾皮质,单腔,直 径小于2cm,不伴有肝囊肿、颅内小动脉瘤等肾外表现。与 ADPKD鉴别困难时,可行基因连锁分析鉴别。
鉴别诊断
➢ 3.结节性硬化症(tuberous sclerosis complex, TSC):又称 Bournerville病,常染色体显性遗传,基因突变率高,影响多器 官细胞移行和分化。主要表现为皮肤损害和中枢神经系统损害, 双肾受累常见,约50%患者出现肾脏血管平滑肌脂肪瘤,30%患者 发现肾囊肿,25%患者发生肾脏恶性肿瘤。
➢ 4.多囊性肾发育不良:肾脏未能正常发育形成囊肿的先天性疾患, 完全性肾发育不良累及双肾者常在新生儿期死亡。累及单侧肾脏者 可无症状,患肾常有肾脏异位,肾脏表面布满囊肿,无泌尿功能, 对侧健肾代偿性肥大,易发生肾盂积水、肾结石及尿路感染。
➢ 5.多房性囊肿:单侧肾脏受累,表现为正常肾脏组织中存在孤立多 房的囊肿,各房之间互不交通,囊肿与肾盂不交通,囊壁有上皮覆 盖,囊肿外未受累的肾组织正常。可根据腹部X线片、尿路造影、B 超等于ADPKD鉴别。
➢ 2.髓质海绵肾(medullary sponge kidney, MSK):少见, 多为散发,个别病例有常染色体显性遗传特征。排泄性尿 路造影的典型表现为肾盏前有刷状条纹和小囊肿,主要表 现为肾结石、感染和反复发作的血尿。
ห้องสมุดไป่ตู้
鉴别诊断
➢ 3.获得性肾囊肿(acquired cystic kidney disease, ACKD): 发生于基础肾脏病为非肾囊肿性疾病的氮质血症期及维持性透析患 者,透析患者发生率与透析时间成正比,透析时间10年以上者发生 率达90%。肾脏囊肿囊液钠与血清钠比值约等于1,而囊内肌酐与血 肌酐比值>1,可与ADPKD鉴别。
男女患病机会无明显差异; 具有常染色体显性遗传特征,代代发病; 50%病例可发展为终末期肾衰竭; 基因缺陷引起的临床异常除了累积肾脏外,还伴有肝囊肿、 胰腺囊肿、 卵巢囊肿、血管系统及心脏瓣膜结构的畸形。
发病机制
临床诊断
ADPKD的临床表现包括肾脏表现和肾外表现。 ➢ 常见临床表现:
疼痛:腰背部或胁腹部疼痛常见,表现为持续或间歇性 钝痛、胀痛或针刺样疼痛。
➢ 常见并发症:泌尿系感染和结石。 ➢ 肾外表现:肝、脾、胰腺、卵巢囊肿形成,肝囊肿最常见,
及心脏瓣膜异常、结肠憩室、颅内动脉瘤等非囊性病变。
成人多囊肾
多囊肾合并胰腺囊肿
多囊肾合并多囊肝
临床诊断
ADPKD的临床诊断标准包括主要标准和次要标准。 ➢ 主要标准:1.肾脏皮质弥漫散布充满液体的囊肿;2.
➢ 4.脑视网膜血管瘤病(von Hipple-Lindau病,VHL):常染色体 显性遗传,特征是全身多脏器发生肿瘤或囊肿。约76%的VHL患者 有双肾多发囊肿,呈局灶结节样增生,常伴发肾脏实体肿瘤、视 神经或中枢神经瘤,可与ADPKD鉴别。
➢ 5.I型口-面-指综合征(orofaciodigital symdrome type 1): X连锁显性疾病,主要表现为面部畸形、口腔异常及骨骼畸形的 先天性综合征。常累及肾脏,表现为多囊肾,肾外表现可与 ADPKD鉴别。
基因定位
目前已知ADPKD突变基因主要有两个,命名为PKD1和PKD2, 分别占ADPKD患者的85%和15%。第3个基因(PKD3)可能存在, 仅在几个家族中发现,尚未在染色体上定位和克隆。
基因 PKD1
ADPKD基因,转录及翻译产物
染色体定 位
16p3.3
基因长度 (Kb)
52
外显子数 目 46
转录产物 翻译产物 分子量大
(Kb)
小
14
Polycystin-1 460,000
成人多囊肾
概述
➢ 多囊肾病(polycystic kidney disease, PKD)是一种常见的遗传性肾脏病。 主要表现为双侧肾脏肾小球囊或多 个肾小管节段进行性扩张,形成多 个大小不一的液性囊肿,囊肿进行 性增大导致肾脏结构和功能受到破 坏,引起不同程度的肾损害,最终 导致终末期肾衰竭。
➢ 多囊肾病根据遗传方式可分为常染 色体显性遗传多囊肾病(autosomal
鉴别诊断
需要鉴别的遗传性囊性肾病包括:
➢ 1.常染色体隐性多囊肾病(ARPKD):基因定位于6号 染色体。多于婴儿期发病,表现为双侧肾脏明显增大, 内有集合管扩张形成的大小较均一的棱形囊肿。进入 成年后,可通过肝脏超声、肝活检、突变基因检测和 基因连锁分析技术与ADPKD鉴别。
➢ 2.髓质囊性病(medullary cystic disease, MCD): 常染色显性遗传疾病,发病与1号染色体和16号染色体 上的基因相关,发病率低。多于成年起病,一般无肾 外表现。B超、CT检查有助于诊断。
明确的多囊肾病家族遗传史。 ➢ 次要标准:1.多囊肝;2.肾功能衰竭;3.腹壁疝;4.
心脏瓣膜病;5.胰腺囊肿;6.脑动脉瘤;7.精囊腺囊 肿;8.眼睑下垂。
2项主要标准+1项次要标准= ADPKD 第1项主要标准+3项以上次要标准= ADPKD
此外,ADPKD的诊断还需结合家族遗传病史、影像学检
查等方面,B超是诊断ADPKD最常用的方法。