新药Etrasimod(艾曲莫德)合成检索总结报告
药物Elagolix sodium(恶拉戈利钠)合成检索总结报告

药物Elagolix sodium(噁拉戈利钠)合成检索总结报告一、Elagolix sodium(噁拉戈利钠)简介Elagolix sodium(噁拉戈利钠)于2018年7月在美国上市,主要用于治疗子宫内膜异位引起的中度至重度疼痛。
Elagolix sodium(噁拉戈利钠)是一种促性腺激素释放激素受体拮抗剂,常见的不良反应有潮热和盗汗、头痛、恶心、失眠、闭经、焦虑、关节痛、抑郁症相关的不良反应和情绪变化。
Elagolix sodium(噁拉戈利钠)分子结构式如下:CAS:832720-36-2英文名称:Elagolix sodium中文名称:噁拉戈利钠本文主要对Elagolix sodium(噁拉戈利钠)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Elagolix sodium(噁拉戈利钠)合成路线(注:关键中间体其他合成方法请参考下面的总结报告)三、Elagolix sodium (噁拉戈利钠)合成检索总结报告(一)Elagolix sodium (噁拉戈利钠)中间体2的合成方法一合成方法实验步骤参考文献操作方法一In a 100mlreaction bottle,add 3.4g of 1,20ml of water,4.23g of urea,and hydrochloric acid (12mol/L,2ml),and reacted under reflux for 3h,the reaction solution was cooled to 4°C with an ice water bath,and filtered to obtained 3.78g of a white solid 2(yield 91%).CN110498771;(2019);(A);CN110498770;(2019);(A)操作方法二To 2-fluoro-6-(trifluoromethyl)benzylamine 1(51.5g,0.267mmol)in a flask,urea (64g,1.07mmol),HCl (CONC.,30.9mmol,0.374mmol)and water (111mL)were added.The mixture was refluxed for 6hours.The mixture was cooled to ambient temperature,further cooled with ice and filtered to give a yellow solid.Recrystallization with 400mL of EtOAc gave 2as a white solid (46.2g,0.196mmol).WO2005/7164;(2005);(A1);WO2005/7165;(2005);(A1);WO2005/7633;(2005);(A1);WO2005/113516;(2005);(A1).(二)Elagolix sodium (噁拉戈利钠)中间体2的合成方法二合成方法实验步骤参考文献操作方法一Dissolve the above crude compound 3in 40mL of tetrahydrofuran in a 250mL three-necked flask.The system was cooled to -10to 10°C,and after stirring for 0.5h,excess ammonia gas was introduced,and after reacting at room temperature for 12hours,The reaction was quenched by slow dropwise addition of water,and concentrated at 40to 50°C until no significant fraction was obtained.Adding ethyl acetate to extract,washing,drying and concentrating to a large amount of solid precipitation and adding n -heptane toCN110041232;(2019);(A)。
新药Vafidemstat(伐德司他、伐菲德司他)合成检索总结报告

新药Vafidemstat(伐德司他、伐菲德司他)合成检索总结报告一、Vafidemstat(伐德司他、伐菲德司他)2020年4月24日宣布该药已获得西班牙药物管理局(AEMPS)的批准,以开展vafidemstat治疗重症COVID-19患者的II期临床试验。
该项II期临床试验(ESCAPE研究)是一项开放标签、随机II 期临床试验,旨在评估vafidemstat与标准护理治疗相结合治疗重症COVID-19的有效性和耐受性。
Vafidemstat(伐德司他、伐菲德司他)分子结构式如下:英文名称:Vafidemstat中文名称:伐德司他、伐菲德司他本文主要对Vafidemstat(伐德司他、伐菲德司他)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Vafidemstat(伐德司他、伐菲德司他)合成路线三、Vafidemstat (伐德司他、伐菲德司他)合成检索总结报告(一)Vafidemstat (伐德司他、伐菲德司他)中间体2的合成合成方法实验步骤参考文献操作方法一To a solution of diethyl oxalate 1(30g,20mmol)in EtOH (50mL)was added hydrazine hydrate (8.1mL)in EtOH (20mL)drop wise at -20o C.The reaction mixture was stirred at -20o C for 0.5h and filtered.To filtrate was added water (15mL)and cyanogen bromide (16.5g,l64mmol)at rt.The reaction mixture was stirred at rt for 1h.The precipitated solid was filtered,washed with Et 2O (l00mL)and dried under vacuum to afford ethyl 5-amino-i ,3,4-oxadiazole-2-carboxylate 2as a white solid.Yield:10g (31%).WO2018/37223;(2018);(A1)English(二)Vafidemstat (伐德司他、伐菲德司他)中间体3的合成合成方法实验步骤参考文献操作方法一A mixture of 5-amino-l,3,4-oxadiazole-2-carboxylic acid ethyl ester 2(2.0g,12.7mmol),Boc anhydride (4.17g,19.1mmol),DIPEA (1.8mL,14.0mmol),DMAP (413mg,3.2mmol)and DMF (15mL)was stirred at 40°C overnight.The DMF was removed in vacuo and the residue taken up in ethyl acetate (40mL).The reaction was washed with brine (2×100mL),saturated sodium bicarbonate (100mL),0.01M HCl (100mL),and dried over sodium sulfate.The organic phase was concentrated and the residue was purified by chromatography on a silica gel column eluted with Hexane/EtOAc (3:1)to afford Compound 3(2.2g,67%)as a white solid.WO2010/144338;(2010);(A1)English Sodium hydride (280mg,0.007mol)in DMF (10mL)was操作方法二added to a suspension ofEthyl 5-amino-l,3,4-oxadiazole-2-carboxylate 2(1g,0.006mol)in DMF (2mL)at 0°C,stirred for 10mins,then Di-tert-butyl dicarbonate (1.65g,0.0076mol)was added and stirred at RT for 16h.After completion,the reaction mixture was poured into ice water (25mL)and extracted with EtOAc (3×25mL).The combined extracts were washed with cold water (2×25mL),brine (25mL),dried over anhydrous Na 2SO 4,filtered and evaporated.The crude residue was purified by column chromatography (SiO 2)using EtOAc:Petroleum ether (1:3)as eluent to get Ethyl 5-((tert-butoxycarbonyl)amino)-l,3,4-oxadiazole-2-carboxylate 3(900mg,56.2%)as a white solid.WO2012/13728;(2012);(A1)English(三)Vafidemstat (伐德司他、伐菲德司他)中间体4的合成合成方法实验步骤参考文献操作方法一NaBH 4(330mg,0.0087mol)was added to a solution of Ethyl 5-((tert-butoxycarbonyl)amino)-l ,3,4-oxadiazole-2-carboxylate 3(900mg,0.0035mol)in THF (18mL)at 0°C and stirred at RT for 16h.After completion,the solvent was evaporated and the residue was taken in water (15mL)and extracted with EtOAc (3×20mL).The combined extracts were washed with water (20mL),brine (20mL),dried over anhydrous Na 2SO 4,filtered and evaporated.The crude residue was purified by column chromatography (SiO 2)using EtOAc:Petroleum ether (8:2)as eluent to get tert-butyl (5-(hydroxymethyl)-l ,3,4-oxadiazol-2-yl)carbamate 4(450mg,54.2%)as a white solid.WO2012/13728;(2012);(A1)English (四)Vafidemstat (伐德司他、伐菲德司他)中间体5的合成合成方法实验步骤参考文献操作方法一MnO 2(500mg)was added to a solution of tert-butyl (5-(hydroxymethyl)-l,3,4-oxadiazol-2-yl)carbamate (Intermediate 4,450mg,0.0021mol)in THF (9mL)at RT and stirred for 16h.After completion,the reaction mixture was filtered through a pad of celite and the filtrate was evaporated to get crude tert-butyl (5-formyl-l,3,4-oxadiazol -2-yl)carbamate 5(250mg).This crude was carried to next step without further purification.WO2012/13728;(2012);(A1)English。
药物Eravacycline(依拉环素)合成检索总结报告

药物Eravacycline(依拉环素)合成检索总结报告一、Eravacycline(依拉环素)简介Eravacycline(依拉环素)是由Tetraphase Pharms公司研发,并于2018年8月在美国上市,主要用于治疗18岁及以上患者的复杂腹腔内感染。
Eravacycline(依拉环素)作用机制是与30S核糖亚基结合,从而防止了氨基酸残基在延长肽链中的结合,破坏细菌蛋白的合成。
Eravacycline(依拉环素)不良反应:输液部位反应,恶心和呕吐。
Eravacycline(依拉环素)分子结构式如下:CAS:1207283-85-9CAS:1334714-66-7英文名称:Eravacycline英文名称:Eravacycline dihydrochloride 中文名称:依拉环素中文名称:依拉环素盐酸盐二、Eravacycline(依拉环素)合成路线三、Eravacycline (依拉环素)合成检索总结报告(一)Eravacycline (依拉环素)中间体3的合成合成方法实验步骤参考文献合成方法一2,6-Lutidine (75.0µL,0.640mmol,5.0equiv)and tert -butyldimethylsilyl trifluoromethanesulfonate 2(88.0µL,0.380mmol,3.0equiv)were added in sequence to a solution of the cyclohexenone 1(47.0mg,0.130mmol,1.0equiv)in dichloromethane (3mL)at 23°C.The mixture was stirred at 23°C for 3h,then an aqueous potassium phosphate buffer solution (pH 7.0,0.2M,15mL)was added.The biphasic mixture was extracted with dichloromethane (2×20mL)and the organic extracts were combined and dried over anhydrous sodium sulfate.The dried solution was filtered and the filtrate was concentrated,affording the silyl-cyclohexenone 3as a white crystalline solid (56.0mg,91%).WO2005/112945;(2005);(A2)EnglishA 100-mL,single-necked,round-bottomed flask equipped with a Teflon-coated magnetic stirring bar was flushed with argon.The flask was charged with a solution of alcohol 1(1.46g,3.96mmol,1equiv)in tetrahydrofuran (10mL),methanol (10mL),and a 2M aqueous sodium dihydrogen phosphate solution (6mL).The resulting biphasic mixture was degassed by bubbling with argon for 30min.The mixture was stirred at 52o C for 15h.The flask was removed from the heating bath and the product mixture allowed to cool to 23o C,then water (15mL),dipotassium hydrogenphosphate (7.83g),and dichloromethane (40mL)合成方法二was added and the resulting biphasic mixture was stirred for10min.The layers were separated.The aqueous layer wasextracted with two40-mL portions of dichloromethane.Theorganic layers were combined and the combined layers weredried over sodium sulfate.The dried solution was filteredand the filtrate was concentrated to provide a purple residuethat was used directly in the next step.A100-mL,single-necked,round-bottomed flask equipped with aTeflon-coated magnetic stirring bar was flame-dried,thenallowed to cool to23o C under argon.The flask was chargedwith a solution of the product obtained above indichloromethane(20mL).The solution was cooled to0°C,whereupon2,6-Lutidine(0.813mL,7.14mmol,1.8equiv)and tert-butyldimethylsilyl trifluoromethanesulfonate2(1.28mL,5.56mmol,1.4equiv)were added sequentiallydropwise by syringe.The reaction mixture was stirred for15min at0o C,then the cooling bath was removed.The reactionsolution was stirred for20min at23o C,then was partitionedbetween aqueous potassium phosphate buffer solution(pH7,0.05M,40mL)and dichloromethane(35mL).The layerswere separated.The aqueous layer was extracted with one40-mL portion of dichloromethane.The organic layers werecombined and the combined layers were dried over sodiumsulfate.The dried solution was filtered and the filtrate was concentrated.The residue was purified by flash-column chromatography on silica gel(100%dichloromethane,grading to2%ethyl acetate-dichloromethane)to provide theenone3(990mg,50%yield over3steps)as a light-yellowfoam.WO2010/126607;(2010);(A2)English合成方法三A3-L,one-necked,round-bottomed flask was equipped witha Teflon-coated magnetic stirbar.The system wasflame-dried and flushed with argon.The flask was chargedwith a solution of the compound1in dichloromethane(1.45L).The solution was cooled to0o C in an ice bath.2,6-Lutidine(33.5mL,305mmol,2.10equiv)andtert-butyldimethylsilyl trifluoromethanesulfonate2(53.3mL,232mmol,1.6equiv)were added sequentially to thecooled solution.The reaction solution was stirred at0o C for15min and then the cooling bath was removed.The reactionsolution was stirred at23o C for20min,and then waspartitioned between aqueous potassium phosphate buffer(pH7.0,0.05M,1L)and dichloromethane(500mL).Theaqueous layer was separated and further extracted withdichloromethane(500mL).The organic layers wereWO2008/127361;(2008);(A2)English。
药物Netarsudil(奈妥舒迪)合成检索总结报告

药物Netarsudil(奈妥舒迪)合成检索总结报告一、Netarsudil(奈妥舒迪)简介Netarsudil(奈妥舒迪)于2017年12月在美国上市,主要用于治疗开角型青光眼或高眼压症患者的眼内压升高。
Netarsudil(奈妥舒迪)不良反应有角膜变性、滴注部位疼痛和结膜出血等等。
Netarsudil(奈妥舒迪)分子结构式如下:CAS:1254032-66-0CAS:1422144-42-0英文名称:Netarsudil英文名称:Netarsudil Mesylate中文名称:奈妥舒迪中文名称:奈妥舒迪甲磺酸盐二、Netarsudil(奈妥舒迪)合成路线三、Netarsudil (奈妥舒迪)合成检索总结报告(一)Netarsudil (奈妥舒迪)中间体3的合成合成方法实验步骤参考文献操作方法一To a solutionof LiHMDS in THF cooled to -78°C was added a cooled solution (-78o C)of methyl 2-(4-((triisopropyl-silyloxy)methyl)phenyl)acetate (1)in THF via syringe.The solution was stirred at -78°C for 30min.Bromo-methyl phthalimide 2was added directly to the anion and the solution stirred for 2h at -78°C.The reaction was then poured into NH 4Cl(sat)and extracted with EtOAc.The organics were dried (MgSO 4),filtered,and evaporated.Column chromatography (SiO 2,0-20%EtOAc/Hexanes)gave pure methyl 3-(1,3-dioxoisoindolin-2-yl)-2-(4-((triisopropylsilyloxy)methyl)phenyl)propanoate (3).WO2010/127329;(2010);(A1)English (二)Netarsudil (奈妥舒迪)中间体4的合成合成方法实验步骤参考文献操作方法一To methyl 3-(1,3-dioxoisoindolin-2-yl)-2-(4-((triisopropyl-silyloxy)methyl)phenyl)propanoate (3)in THF/H 2O was added LiOH-H2O,and the solution was stirred for 1.5h or until conversion to product was visible by LC-MS.The solution was then poured into EtOAc/NH 4Cl(sat)/1N HCI (3:1)and the aqueous layer was further extracted with EtOAc.The organics were dried (MgSO 4),filtered,and evaporated to give crude 2-(2-carboxy-2-(4-((triisopropylsilyloxy)methyl)phenyl)ethylcarbamoyl)benzoiWO2010/127329;(2010);(A1)Englishc acid (4).(三)Netarsudil (奈妥舒迪)中间体6的合成合成方法实验步骤参考文献操作方法一To 2-(2-carboxy-2-(4-((triisopropylsilyloxy)methyl)phenyl)ethylcarbamoyl)benzoicacid (4)in pyridine was added EDC,DMAP and 6-aminoisoquinoline 5and the solution was flushed with N 2,capped,and stirred overnight.The mixture was poured into EtOAc/NaHCO 3(sat)and the aqueous layer was further extracted with EtOAc.The organics were dried (MgSO 4),filtered,and evaporated.Column chromatography (SiO 2,5%MeOH/CH 2Cl 2)gave pure 3-(1,3-dioxoisoindolin -2-yl)-N-(isoquinolin-6-yl)-2-(4-((triisopropylsilyloxy)-methyl)phenyl)propanamide (6).WO2010/127329;(2010);(A1)English (四)Netarsudil (奈妥舒迪)中间体7的合成合成方法实验步骤参考文献操作方法一To 3-(1,3-dioxoisoindolin-2-yl)-N-(isoquinolin-6-yl)-2-(4-((triisopropylsilyloxy)methyl)phenyl)propanamide (6)in EtOH was added NH 2-NH 2and the solution was refluxed for 1.2-2h.The solids were filtered,and the solvents were evaporated.Column chromatography (SiO 2,5%2N NH 3-MeOH/CH 2Cl 2)gave pure 3-amino-N-(isoquinolin-6-yl)-2-(4-((triisopropylsilyloxy)methyl)phenyl)propanamide (7).WO2010/127329;(2010);(A1)English(五)Netarsudil (奈妥舒迪)中间体8的合成。
药物Letermovir(莱特莫韦)合成检索总结报告
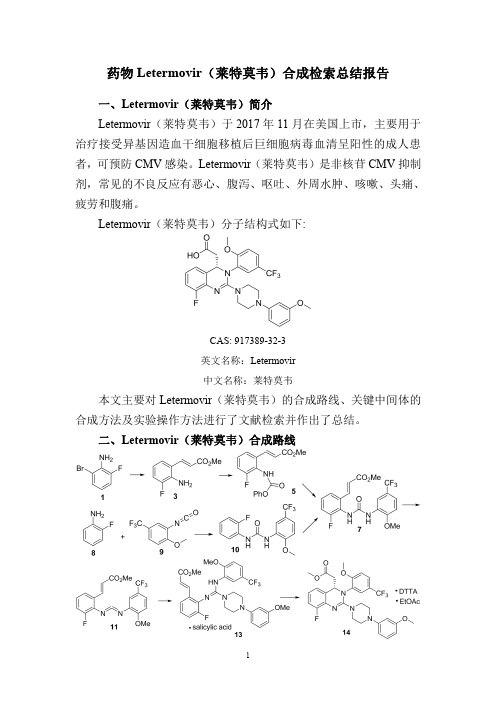
药物Letermovir(莱特莫韦)合成检索总结报告一、Letermovir(莱特莫韦)简介Letermovir(莱特莫韦)于2017年11月在美国上市,主要用于治疗接受异基因造血干细胞移植后巨细胞病毒血清呈阳性的成人患者,可预防CMV感染。
Letermovir(莱特莫韦)是非核苷CMV抑制剂,常见的不良反应有恶心、腹泻、呕吐、外周水肿、咳嗽、头痛、疲劳和腹痛。
Letermovir(莱特莫韦)分子结构式如下:CAS:917389-32-3英文名称:Letermovir中文名称:莱特莫韦本文主要对Letermovir(莱特莫韦)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Letermovir(莱特莫韦)合成路线三、Letermovir (莱特莫韦)合成检索总结报告(一)Letermovir (莱特莫韦)中间体3的合成合成方法实验步骤参考文献操作方法一To adegassed solution of 2-bromo-6-fluoroaniline 1(1,99.5g,0.524mol),methyl acrylate 2(95.0mL,1.05mol),Chloro[(tri-tert-butylphosphine)-2-(2-aminobiphenyl)]palladium(II)(0.537g,1.05mmol)in isopropyl acetate (796mL),was added degassed N,N-dicyclohexylmethylamine (135mL,0.628mol).The resulting reaction was heated to 80°C and allowed to stir at this temperature for 5hours.The resulting slurry was cooled to 20°C and filtered.The filtrate was washed with 1M citric acid to provide a solution that contained compound 3(99.3g,97%assay yield)in isopropyl acrylate,which was used without further purification.WO2015/88931;(2015);(A1)English (二)Letermovir (莱特莫韦)中间体5的合成合成方法实验步骤参考文献操作方法一To a solution of compound 3(48.8g,0.250mol)in 683mL of isopropyl acetate was added 244mL of water,followed by di-sodium hydrogen phosphate (53.2g,0.375mol).To the resulting solution was added phenyl chloroformate 4(39.2mL,0.313mol)dropwise over 30minutes.The resulting reaction was heated to 30°C and allowed to stir at this temperature for 5hours for 4hours and then was heated to 60°C and allowed to stir at this temperature for 5hours for an additional 2hours to remove excess phenyl chloroformate.An additional 293mL of isopropyl acetate was then added and the reaction mixture was allowed to stirWO2015/88931;(2015);(A1)Englishat room temperature until the solids completely dissolvedinto solution.The resulting reaction mixture was transferred to a separatory funnel and the organic phase was washed with 98mL of water and collected to provide a solution of compound 5in isopropyl acetate,which was used without further purification.操作方法二To a stirred crude i -PrOAc solution of acrylate 3(15.9kg,8.2wt %15,6.66mol)was added H 2O (6.50L)and Na 2HPO 4(1.42kg,10.0mol)followed by phenyl chloroformate 4(1.04L,8.33mol)dropwise over 30min.The reaction mixture was stirred at RT for 12h before being heated to 60°C and stirred for a further 2h.The mixture was then diluted with i -PrOAc (7.00L)and held at 60°C until all solids were dissolved.The aqueous phase was then separated and the organics washed with H 2O (2.60L)before being cooled to RT to afford a crude i-PrOAc slurry of carbamate 5(ca.95%assay yield)which was used directly without further purifification.For the purposes of characterization,a sample of pure carbamate 5was isolated by crystallization from a sample of the crude solution diluted with heptane.Mp 154−158°anic Process Research and Development ;vol.20;nb.6;(2016);p.1097–1103.(三)Letermovir (莱特莫韦)中间体7的合成方法一合成方法实验步骤参考文献操作方法一A solution of compound 5(79.0g,0.250mol),2-methoxy-5-(trifluoromethyl)aniline 6(52.7g,0.276mol),and 4-dimethylaminopyridine (0.92g,0.0075mol)in isopropyl acetate (780mL)was heated to reflux and allowed to stir at this temperature for 5hours.The resulting slurry was cooled to 20°C,then allowed to stir at this temperature for for two hours at this temperature,then filtered.The collected filter cake was dried in vacuo to provide compound 7(95.0g,0.230mol)as a white solid,which was used without further purification.WO2015/88931;(2015);(A1)English 操作方法To a stirred crude i-PrOAc solution of carbamate 5(<6.66mol)was added 2-methoxy-5-(triflfluoromethyl)aniline 6(1.40kg,7.33mol).The slurry was diluted with i -PrOAc,heated to 40−45°C,and a constant volume distillation carried out to azeotropically dry the mixture (KF <400ppm).Organic Process Research and Development ;。
抗偏头痛药阿莫曲坦的合成工艺研究

抗偏头痛药阿莫曲坦的合成工艺研究一、前言选题的依据和意义阿莫曲坦是新型的第二代选择性5-HT1B/1D受体激动剂,是一种偏头痛急性发作的有效医治药,有很高的选择性,副作用小。
研究优化它的合成工艺,具有显著的社会意义和经济价值。
1.2国内外研究综述偏头痛是一种慢性多发病,通过对其发病机理的研究,已开发出5-HT受体激动剂(即曲坦类药物)作为偏头痛急性发作的有效医治药。
阿莫曲坦是经结构改造开发的新型第二代选择性5-HT1B/1D受体激动剂,对颅内血管的5-HT1B/1D受体的亲和力有很高的选择性,对冠状动脉的致痉作用较其他曲坦类药物小,克服了第一代曲坦类药物如舒马曲坦的某些不良反映,尤其是胸痛发作率明显减少,对人脑动脉作用比舒马曲坦强25倍,是对成年中重度急性发作偏头痛的最新型的有效医治药物。
制备阿莫曲坦的方式有多种,一些文献报导的合成线路如下:一、 US 5565447中描述了利用氧化铜催化剂并用喹啉作为溶剂,通过中间体1-[[2-羧基-3-(二甲氨基乙基)-5-吲哚]甲磺酰]吡咯烷的脱羧作用来制备3,5-二取代的吲哚衍生物,如阿莫曲坦。
该法影响产物的总产率及阿莫曲坦的质量。
二、Tetrahedron,2001,第57卷,第1041-1047页中报导了由Heck环化来制备吲哚环。
持续的方式包括多个步骤。
用溴处置1-(4-氨基-苯甲基磺酰基)吡咯烷,然后用三氟乙酸处置,以便在2-位引入含溴部份和苯胺氮保护。
另外,利用LDA和4-溴代巴豆酸甲酯进行烯丙基化。
利用Pb(OAc)2实现了Heck环化。
所取得的吲哚-3-醋酸酯被水解为相应的酸,然后相应的酸被转化为酰氯,并通过在碱性介质中与二甲胺反映进一步转化为二甲基酰胺。
最后,酰胺羰基的还原产生了所需化合物。
此法阿莫曲坦总产率超级低。
3、 ES 2,084,560描述了基于Fischer吲哚合成制备阿莫曲坦的方式,其利用苯肼和4-氯-丁醛二乙基缩醛生成1-[[3-(2-氨基乙基)-5-吲哚] 甲磺酰]吡咯烷。
omacor临床试验总结翻译
关于浓缩型Omega-3多不饱和脂肪酸—Omacor的临床综述医学博士哈罗德·贝斯美国肯塔基州路易斯维尔市L-MARC研究中心再版地址:肯塔基州路易斯维尔市伊利诺斯大街3288号,L-MARC研究中心,医学博士哈罗德·贝斯邮编:40213电子邮件地址:HBaysMD@.0002-9149/06/$-详见扉页© 2006 爱思唯尔公司版权所有。
网址:标识符:10.1016/j.amjcard.2005.12.029Omacor(ω-3-酸乙酯;由新泽西州自由角信赖制药公司生产)为一种经高度提纯的处方型ω-3脂肪酸,每粒容量为1g的胶囊中含高浓度二十碳五烯酸(EPA)(465mg)、二十二碳六烯酸(DHA)(375mg)及4mg(6 IU)维生素E。
在日服用4粒胶囊的典型剂量中,无论是作为单一治疗药物还是配合羟甲基戊二酸单酰辅酶A(HMG-CoA)还原酶抑制剂(他汀类)或贝特类药物使用,Omacor均具有显著降低血浆甘油三酯水平的功效。
与此同时,Omacor还可以适当提高低密度脂蛋白胆固醇和高密度脂蛋白胆固醇的血浆水平,并对脂蛋白的粒子大小及子类分布产生有益影响。
Omacor具有良好的耐受性,除出现轻微胃肠道症状外并无其他副作用(至今未有报告称服用该药物会导致高血糖、异常出血、肌酶和肝酶含量升高以及肾功能或神经功能异常)。
经过密集的提纯工序,Omacor的“鱼腥味”已降至最低,且未有报告称暴露在环境毒素中的Omacor 会引发维生素过多症或其他疾病。
Omacor无疑为治疗高甘油三脂血症提供了一种安全、有效、耐受性高的途径。
© 2006爱思唯尔公司版权所有。
(美国心脏病学杂志2006;98[增刊]:71i-76i)美国心脏协会(AHA)建议患有冠状动脉疾病(CAD)的患者每日混合服用剂量超过1g的十二碳五烯酸(EPA)和二十二碳六烯酸(DHA),同时建议血浆甘油三酯(TG)水平偏高的患者每日服用2至4g上述混合药物(表1)。
新药Tepotinib(特泊替尼)合成检索总结报告
新药Tepotinib(特泊替尼)合成检索总结报告一、Tepotinib(特泊替尼)简介2019年09月12日,美国食品和药物管理局已授予其靶向抗癌药MET抑制剂Tepotinib(特泊替尼)突破性药物资格,用于治疗接受含铂化疗后病情进展、携带MET基因第14号外显子跳跃突变的转移性非小细胞肺癌患者。
2018年3月,Tepotinib(特泊替尼)被日本卫生劳动福利部授予了治疗携带MET基因第14号外显子跳跃突变的晚期NSCLC患者的SAKIGAKE资格(创新药物)。
Tepotinib(特泊替尼)分子结构式如下:英文名称:Tepotinib中文名称:特泊替尼本文主要对Tepotinib(特泊替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Tepotinib(特泊替尼)合成路线三、Tepotinib (特泊替尼)合成检索总结报告(一)Tepotinib (特泊替尼)中间体3的合成合成方法实验步骤参考文献操作方法一2.2L of a freshly prepared 1.5M sodium methoxide solution are added dropwise with stirring to a suspension of 259g (1.09mol)of 3-methoxycarbonylbenzamidinium acetate 1and 528g (1.08mol)of ({2-dimethylamino-1-[dimethyli-mmoniomethyl]vinylamino}methylene)dimethyl-ammonium dihexafluorophosphate 2(“a minoreductone precursor”,prepared in accordance with C.B.Dousson et al.,Synthesis 2005,1817)in 1L of methanol.The reaction mixture is then warmed to 60°C.over the course of 40min and held at this temperature for 30min.The reaction mixture is then cooled to room temperature,diluted with 10L of dichloromethane and washed three times with 5L of water each time.The organic phase is dried over sodium sulfate and evaporated.The residue is recrystallised from ethyl acetate:methyl 3-[5-(dimethylaminomethyleneamino)pyrimidin-2-yl]-benzo ate 3as beige crystals;m.p.146°2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1)English;US2011/269765;(2011);(A1)English;US2011/269756;(2011);(A1)English 操作方法二100g of 3-hydroxymethylbenzamidinium acetate 1(419.75mmol)and 204.93g of a minoreductone precursor 2(419.74mmol)are suspended in 1000ml of dried MeOH in an N 2-flushed 2L three-necked flask,and a freshly prepared solution of 28.99g of sodium in 300ml of MeOH is added dropwise with stirring,and the mixture is subsequently stirred at 60°C.for 30min,giving a clear solution.For work-up,the reaction batch is cooled,diluted with dichloromethane,washed 2×with water,dried over sodium sulfate and evaporated to dryness in a rotary evaporator.The residue 3is crystallised from a little methanol and diethyl 2010/311733;(2010);(A1)English(二)Tepotinib (特泊替尼)中间体4的合成合成方法实验步骤参考文献操作方法一160ml(2.88mol)of concentrated sulfuric acid are added to a suspension of 103.5g (364mmol)of methyl 3-[5-(di-methylaminomethyleneamino)-pyrimidin-2-yl]benzoate 3in 1.3L of water,and the mixture is heated at the boil for 4hours.The reaction mixture is cooled to room temperature,diluted with water and filtered with suction.The residue is washed with water and dried in vacuo:3-(5-hydroxypyri-midin-2-yl)benzoic acid as brownish crystals;m.p.293-295°2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1);US2011/269765;(2011);(A1);US2011/269756;(2011);(A1).操作方法二103.5g of methyl 3-[5-(dimethylaminomethylenamino)-pyrimidin-2-yl]benzoate 3(364.04mmol)are suspended in 1300ml of water in a 2l single-necked flask,and 160ml of conc.sulfuric acid (95-97%)(2.88mol)are subsequently added,and the reaction batch is warmed at 130°C.(oil-bath temperature)for 4h.For work-up,the reaction batch is cooled,and the precipitate formed is filtered off,washed with water and dried at 50°C.in a vacuum drying cabinet.Yield:78.9g (364.5mmol)of 3-(5-hydroxypyrimidin-2-yl)-benzoic acid 2010/311733;(2010);(A1)English(三)Tepotinib (特泊替尼)中间体5的合成合成方法实验步骤参考文献操作方法一32.7ml (445mmol)of thionyl chloride are added to a suspension of 88.0g (366mmol)of 3-(5-hydroxypyrimidin -2-yl)benzoic acid 4in 1.4l of methanol,and the mixture is heated at 80°C.for 2hours.20ml (276mmol)of thionyl chloride and,after 2hours,a further 10ml (138mmol)of thionyl chloride are then added.After each addition,the reaction mixture is stirred at 80°C.for 2hours.The reaction mixture is concentrated to a volume of about 300ml in vacuo.The resultant precipitate is filtered off and dried in vacuo:methyl 3-(5-hydroxypyrimidin-2-yl)benzoate 5as brownish crystals;m.p.219-223°C.US2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1)English;US2011/269765;(2011);(A1)English;US2011/269756;(2011);(A1).78.8g of 3-(5-hydroxypyrimidin-2-yl)benzoic acid 4are suspended in 1.4l of absolute methanol,and 32.7ml of。
上市新药Siponimod(辛波莫德)合成检索总结报告
上市新药Siponimod(辛波莫德)合成检索总结报告一、Siponimod(辛波莫德)简介Siponimod(辛波莫德)是由诺华公司研发,2019年3月26日在美国上市,主要用于复发型多发性硬化症(MS)成人患者的治疗,包括临床孤立综合征(CIS)、复发缓解型多发性硬化症(RRMS)和活动性继发进展型多发性硬化症(SPMS)。
Siponimod(辛波莫德)是一种鞘氨醇-1-磷酸(S1P)受体调节剂。
与SlP受体1和5具有高亲和力,阻止淋巴细胞从淋巴结流出,从而减少了外周血中的淋巴细胞数量。
Siponimod(辛波莫德)不良反应:头痛,高血压和转氨酶升高。
Siponimod(辛波莫德)分子结构式如下:CAS:1230487-00-9英文名称:Siponimod中文名称:辛波莫德化学名称:3-[[4-[(1E)-1-[[[4-环己基-3-(三氟甲基)苯基]甲氧基]亚氨基]乙基]-2-乙基苯基]-甲基]-3-氮杂环丁烷羧酸富马酸盐(2:1)二、Siponimod(辛波莫德)合成路线三、Siponimod(辛波莫德)合成检索总结报告(一)Siponimod(辛波莫德)中间体3的合成序号实验步骤参考文献1To a solution of ethyl N-hydroxyacetimidate2(3.0g,29.3mmol)in dry DMF (25mL)was added KOBu-t (3.3g,29.3mmol)and the mixture was stirred at room temperature for 20min.A solution of benzyl bromide 1(8.0g,29.3mmol)in dry DMF (5mL)was then added.The resulting mixture was stirred at room temperature for 5h.The reaction mixture was partitioned in H 2O (200mL)and 20%EtOAc/hexane (100mL).After separation,the aqueous layer was further extracted with 20%EtOAc/hexane (2X50mL).The combined organic layer was washed with brine and dried over anhydrous Na 2SO 4.After concentration,the residue was passed through a silica gel pad (a depth of 3cm)in a filtration funnel and washed with 10%EtOAc/hexane.The desired product 3was obtained after concentration as a colorless liquid (8.60g,100%).ACS Medicinal Chemistry Letters ;vol.4;nb.3;(2013);p.333-337(二)Siponimod (辛波莫德)中间体4的合成序号实验步骤参考文献1Anhydrous ZnCl 2(7.96g,58.4mmol)was dissolved in dry degassed NMP (25mL)in a 2-neck flask while heated at 100o C under argon and the resulting solution was allowed to cool to room temperature.One neck of the flask was connected with a distillation set-up while the other was sealed with septa.To the above ZnCl 2solution was added cyclohexyl magnesium chloride (2M in Et 2O,26.5mL,53mmol)via syringe.The reaction was exothermic and Et 2O was evaporated.After the completion of addition,the viscous mixture was stirred at room temperature for 5min before elevating the temperature to 80o C to allow for the complete evaporation of Et2O (ca.30min)to give an unstirrable solid.After cooled to room temperature,chloride 3(7.85g,26.5mmol)and Pd(PBut 3)2(0.678g,1.33mmol)were added.The flask was flushed with argon and sealed with septa.The mixture was then heated to 140o C and stirred for 1h.After cooled to room temperature,the mixtureACS Medicinal Chemistry Letters ;vol.4;nb.3;(2013);p.333-337。
新药Oliceridine(奥塞利定)合成检索总结报告
新药Oliceridine(奥塞利定)合成检索总结报告
一、Oliceridine(奥塞利定)简介
Oliceridine(奥塞利定)是选择性阿片μ受体激动剂,只激活G 通路而不影响β-arrestin通路,近期完成的III期临床研究表明等效镇痛剂量下TRV130引起的胃肠功能障碍和呼吸抑制风险比吗啡少,治疗窗比吗啡更宽,具有广阔的临床和市场前景。
Oliceridine(奥塞利定)分子结构式如下:
英文名称:Oliceridine
中文名称:奥塞利定
本文主要对Oliceridine(奥塞利定)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Oliceridine(奥塞利定)合成路线
三、Oliceridine(奥塞利定)合成检索总结报告(一) Oliceridine(奥塞利定)中间体3的合成方法一
(二) Oliceridine(奥塞利定)中间体3的合成方法二
(三) Oliceridine(奥塞利定)中间体5的合成。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
新药Etrasimod(艾曲莫德)合成检索总结报告
一、Etrasimod(艾曲莫德)简介
Etrasimod(艾曲莫德)对特定的免疫细胞类型提供系统性和局部效应,并有可能治疗多种免疫介导的炎症疾病,包括溃疡性结肠炎、克罗恩病、嗜酸性食管炎、特应性皮炎和斑秃。
近日,Arena Pharmaceuticals宣布其Etrasimod用于治疗溃疡性结肠炎的II期临床试验取得积极顶线结果,与安慰剂相比每日口服2mg Etrasimod的患者在所有的主要、次要和临床缓解终点上都表现出统计学上的显著提高,并且安全性数据优秀。
Etrasimod(艾曲莫德)分子结构式如下:
英文名称:Etrasimod
中文名称:艾曲莫德
本文主要对Etrasimod(艾曲莫德)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Etrasimod(艾曲莫德)合成路线
(一) Etrasimod(艾曲莫德)中间体15的合成路线
(二) Etrasimod(艾曲莫德)中间体7的合成路线一
(三) Etrasimod(艾曲莫德)中间体7的合成路线二
(四) Etrasimod(艾曲莫德)的合成路线(从7开始合成)
三、Etrasimod(艾曲莫德)合成检索总结报告
(一) Etrasimod(艾曲莫德)中间体3的合成
(二) Etrasimod(艾曲莫德)中间体4的合成
(三) Etrasimod(艾曲莫德)中间体5的合成
(四) Etrasimod(艾曲莫德)中间体6的合成
(五) Etrasimod(艾曲莫德)中间体7的合成方法一
(六) Etrasimod(艾曲莫德)中间体7的合成方法二①Etrasimod(艾曲莫德)中间体9的合成。