硕士研究生分子生物学实验步骤
分子生物学实验方法与步骤【可编辑全文】

可编辑修改精选全文完整版分子生物学实验方法与步骤表达蛋白的SDS-聚丙烯酰胺凝胶电泳分析一、原理细菌体中含有大量蛋白质,具有不同的电荷和分子量。
强阴离子去污剂SDS与某一还原剂并用,通过加热使蛋白质解离,大量的SDS 结合蛋白质,使其带相同密度的负电荷,在聚丙烯酰胺凝胶电泳(PAGE)上,不同蛋白质的迁移率仅取决于分子量。
采用考马斯亮兰快速染色,可及时观察电泳分离效果。
因而根据预计表达蛋白的分子量,可筛选阳性表达的重组体。
二、试剂准备1、30%储备胶溶液:丙烯酰胺(Acr)29.0g,亚甲双丙烯酰胺(Bis)1.0g,混匀后加ddH2O,37O C溶解,定容至100ml, 棕色瓶存于室温。
2、1.5M Tris-HCl(pH 8.0:Tris 18.17g加ddH2O溶解, 浓盐酸调pH至8.0,定容至100ml。
3、1M Tris-HCl(pH 6.8:Tris 12.11 g加ddH2O溶解, 浓盐酸调pH至6.8,定容至100ml。
4、10% SDS:电泳级SDS 10.0 g加ddH2O 68℃助溶,浓盐酸调至pH 7.2,定容至100ml。
5、10电泳缓冲液(pH 8.3:Tris 3.02 g,甘氨酸18.8 g,10% SDS 10ml加ddH2O溶解, 定容至100ml。
6、10%过硫酸铵(AP): 1gAP加ddH2O至10ml。
7、2SDS电泳上样缓冲液:1M Tris-HCl (pH 6.82.5ml,-巯基乙醇1.0ml,SDS 0.6 g,甘油2.0ml,0.1%溴酚兰1.0ml,ddH2O 3.5ml。
8、考马斯亮兰染色液:考马斯亮兰0.25 g,甲醇225ml,冰醋酸 46ml,ddH2O 225ml。
9、脱色液:甲醇、冰醋酸、ddH2O以3∶1∶6配制而成。
二、操作步骤采用垂直式电泳槽装置(一)聚丙烯酰胺凝胶的配制1、分离胶(10%的配制:ddH2O 4.0ml30%储备胶 3.3ml1.5M Tris-HCl2.5ml10% SDS 0.1ml10% AP 0.1ml取1ml上述混合液,加TEMED(N,N,N’,N’-四甲基乙二胺10μl 封底,余加TEMED4μl ,混匀后灌入玻璃板间,以水封顶,注意使液面平。
分子生物学实验方法与步骤

DNA分子的限制性内切酶消化限制性内切酶可特异地结合于一段被称为限制酶识别序列的DNA序列位点上并在此切割双链DNA。
绝大多数限制性内切酶识别长度为4、5或6个核苷酸且呈二重对称的特异序列,切割位点相对于二重对称轴的位置因酶而异。
一些酶恰在对称轴处同时切割D NA双链而产生带平端的D NA片段,另一些酶则在对称轴两侧相对的位置上分别切断两条链,产生带有单链突出端(即粘端)的DNA片段。
1个单位限制性内切酶是指在最适条件下,在50μl体积1小时内完全切开1μgλ噬菌体DNA所需的酶量。
不同的限制性内切酶生产厂家往往推荐使用截然不同的反应条件,甚至对同一种酶也如此。
但是,几乎所有的生产厂家都对其生产的酶制剂优化过反应条件,因此购买的内切酶说明书上均有其识别序列和切割位点,同时提供有酶切缓冲液(buffer,10×、5×)和最适条件,使得酶切反应变得日益简单。
一、限制性内切酶对D NA消化的一般方案1.限制性内切酶反应一般在灭菌的0.5ml离心管中进行。
2.20μl体积反应体系如下:DNA0.2-1 μg10×酶切buffe r 2.0 μl限制性内切酶1-2 u(单位)加ddH2O至20 μl限制性内切酶最后加入,轻轻混匀,稍加离心,放置于最适温度水浴并按所需的时间温育,一般为37℃水浴消化1hr。
3.用于回收酶切片段时,反应总体积可达50-200μl,各反应组分需相应增加。
4.多种酶消化时若缓冲液条件相同,可同时加入;否则,先做低温或低盐的酶消化,再做高温或高盐的酶消化。
5.酶切结束时,加入0.5M EDTA(pH 8.0)使终浓度达10mM,以终止反应。
或将反应管在65℃水浴放置10m i n以灭活限制性内切酶活性。
硕士分子生物学实验步骤
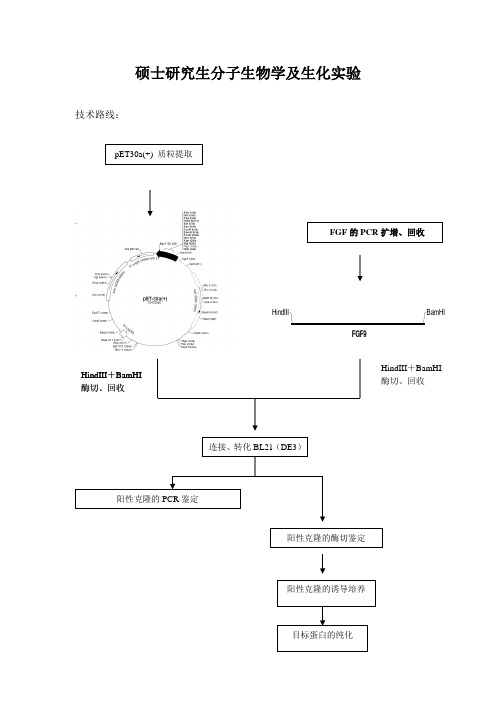
硕士研究生分子生物学及生化实验技术路线:HindIIIBamHIFGF9FGF 的PCR 扩增、回收HindIII 连接、转化BL21(DE3)阳性克隆的PCR 鉴定+BamHI 酶切、回收HindIII +BamHI 酶切、回收pET30a(+) 质粒提取阳性克隆的酶切鉴定阳性克隆的诱导培养 目标蛋白的纯化一、感受态细胞的制备1.实验材料及试剂受体菌:BL21(DE3)、质粒pET-30a+转化液:50mM CaCl2培养基:LB培养基(pH7.0) 121℃,20min 高压灭菌NaCl 5g/L酵母提取物5g/L胰蛋白胨10g/L2..实验步骤1. 感受态细胞的制备1)活化的BL21(DE3)平板上挑取一单菌落,接种于100mL LB液体培养基中37℃振荡培养过夜。
2)取培养过夜的菌液100μL接种于100ml新鲜的LB培养基中,37℃振荡培养约5h。
3)每组取2支10ml离心管,各转入8ml菌液,4000r/min离心3min,收集菌体。
4)弃去上清液,在涡旋器上将细胞悬浮于冰冷的2mL 50mmol/L CaCl2溶液中,将2管合并为1管,并在冰上放置15-30min。
4000r/min离心3min。
5)弃去上清液,加入1ml冰冷的50mmol/L CaCl2溶液,小心悬浮细胞,置冰上,即制成了感受态细胞悬液,可直接用于转化实验,也可加入占总体积20%左右高压灭菌过的甘油,混合后分装于Eppendorf管中,置于-70℃条件下,可保存6-12个月。
二、FGF的PCR扩增与回收(一)试剂电泳缓冲液(50×TAE)Tris 242g冰醋酸 57.1mlEDTA(0.5mol/L pH8.0) 100ml使用时用蒸馏水稀释50倍。
DNA快速纯化/回收试剂盒(二)方法1.PCR反应条件目的基因模板来源于质粒pcDNA3.1-FGF,将质粒稀释100倍,PCR反应采用50μl反应体系。
分子生物学实验步骤

细菌的活化及培养1.将实验室用到的烧杯、锥形瓶、培养皿等实验器皿用超声机超声20min,然后用自来水冲洗,再用蒸馏水冲洗干净,放入烘箱中烘干。
按照培养基成分配置液体培养基,分别调好pH值后,然后进行灭菌30min。
牛肉膏蛋白胨培养基配方接种:在超净工作台中,取1 mL的菌种加入液体培养基中,混匀,37℃恒温振荡培养12h。
2.菌种的保存(采用甘油保藏法)先取800uL的菌悬液于1.5mL的无菌离心管中,再加入200uL的灭菌甘油,用封口膜将离心管口封号,以上操作均在超净工作台中完成,然后将装有甘油和菌悬液的混合液在漩涡震荡以上混匀,保存至-78℃。
细菌DNA的提取采用TIANamp Bacteria DNA kit试剂盒对单增李斯特菌进行基因组DNA抽提。
操作步骤如下:1)取细菌培养液1ml,1000rpm离心一分钟,尽量吸净上清。
2)向菌体沉淀中加入200μl缓冲液GA,振荡至菌体彻底悬浮。
注意:对于较难破壁的革兰氏阳性菌,可略过第二步,加入溶菌酶进行破壁处理,具体方法为:加入180μl缓冲液(110μl TE缓冲液,70μl溶菌酶),37℃处理30min以上。
3)向管中加入20μl蛋白酶K溶液,混匀。
4)加入220μl缓冲液GB(改良磷酸盐缓冲液),振荡15s,70℃放置10min,溶液应变清亮,简短离心以去除管盖内壁的水珠。
注意:加入缓冲液GB时可能会产生白色沉淀,一般70℃放置时会消失,不会影响后续实验。
如溶液未变清亮,说明细胞裂解不彻底,可能导致提取DNA 量少和提取出的DNA不纯。
5)加220μl无水乙醇,充分振荡混匀15s,此时可能出现絮状沉淀,简短离心以去除管盖内壁的水珠。
6)将上一步所得溶液和絮状沉淀都加入一个吸附柱CB3中(吸附柱放入收集管中),12000rpm离心30s,倒掉废液,将吸附柱CB3放入收集管中。
7)向吸附柱CB3中加入500μl缓冲液GD(使用前请先检查是否已加入无水乙醇),12000rpm离心30s,倒掉废液,将吸附柱CB3放入收集管中。
分子生物学实验方案设计(五篇模版)

分子生物学实验方案设计(五篇模版)第一篇:分子生物学实验方案设计梅衣属ITS条形码物种的快速鉴定实验目的:一、学习并熟练地衣DNA提取技术二、掌握PCR技术的原理及操作三、DNA条形码技术的应用实验技术路线:1、地衣总DNA的提取2、PCR扩增3、基因测序4、基因序列对比地衣总DNA提取:1.取带有子囊盘或未有子囊盘的带藻地衣体30~300mg,用无菌水冲洗,除去表面杂质,再用DNA提取液浸泡2~3h(4℃)。
2.将地衣体取出,滤纸吸干表面液体,剪碎放于预冷的研钵中,加入液氮研磨至粉末状。
3.将粉末小心、全部转入1.5ml离心管中,加入400μL DNA提取液,混匀。
4.加入10% SDS 50μL、氯化苄150μL,振荡混合,于50℃保温1h,每隔10min振荡混合一次。
5.保温1h后,每管加入3mol/L NaAc 50μL,混匀冰浴15min.6.用等体积酚:氯仿:异戊醇(25:24:1)和等体积酚:氯仿(1:1)各抽提一次,直至无白色沉淀为止。
7.移上清液于另一离心管,加入2倍体积无水乙醇,4℃放置2~3h,15000r/min,离心10min(4℃),弃上清液。
8.将沉淀用70%乙醇冲洗2次后,真空干燥,再加入50~80μL TE 缓冲液,保存于冰箱(4℃)PCR扩增:稀释50 倍用于后续 PCR 操作。
真菌 ITS rDNA 由通用引物 ITS5(5′-GGAAGTAAAAGTCGTAACAAGG-3′)、ITS4(5′-TCCTCCGCTTATTGATATGC-3′)。
PCR 反应条件:95℃预变性5min;95℃变性30s,52℃退火30s,72℃延伸 2min,反应 32 个循环;72℃延伸 10min,4℃保存。
反应结束后,PCR 产物通过0.8%的琼脂糖凝胶电泳检验,ABI3730DNA 分析仪测序。
序列比对:所获测序结果序列包含小亚基部分结尾序列、ITS1-5.8S-ITS2、大亚基部分起始序列,去除大小亚基部分序列后所得片段大小约500bp,即为所需的序列利用 DNAMAN(Lynnon Biosoft)软件将测序结果同 GenBank 下载数据进行比对分析,如果序列长度不同,则只保留共有区段。
硕士研究生分子生物学实验步骤

• • • •
• •
取质量
质粒DNA的限制性内切酶酶切
• 反应体系如下: 灭菌双蒸水 10×限制酶buffer 质粒DNA BamHІ HindШ 6μl 2μl 10μl 1μl 1μl
总体积20μl,混匀,37℃水浴1-3 h,,用1%琼脂糖凝胶 电泳鉴定结果。
RT-PCR扩增目的基因
第一步:RT(总体积25μl)
Taq酶
2.5μl
二、质粒转化、平板筛选 • 制备含含100μg/ml氨苄青霉素(Amp)的LB琼脂平板 上 (50 ℃ 时加入Amp )
• 取感受态细菌100μl置于无菌EP管中;
• 加入重组质粒DNA10 μl,轻轻旋转混匀,冰上放置 30min,42 ℃热休克90sec(勿超时!),冰浴速冷12min。 • 加入无抗生素的LB培养基400μl,于37 ℃摇床(100150rpm)温和振摇45min,使细菌复苏并表达质粒的抗 性基因。 • 取100μl菌液均匀涂铺于含Amp的LB琼脂平板上 , 37 ℃ 20min后,倒置平板继续培养10-16h(小于20h) • 挑取单菌落置5mlLB培养基中(含Amp) 37 ℃摇床培 养8-12h后,提取质粒,限制酶切鉴定。
RNA模板 2μl Oligo(dT)15
1.5μl
65 ℃,5min; 4 ℃,1min; DEPC处理水 14μl 5xRT buffer 5μl dNTP 1.5μl RT酶 1μl
2000rpm离心20sec后,42 ℃,60min逆转录;85 ℃,5min终 止反应;4 ℃,3m• 离心力要注意不要超过2500g,建议1500-2000g 5min。 • 涂板要注意,不要觉得不匀而多次反复,轻轻在平板上 带一下感觉板上大部分地方走过即可,特别在冰箱中保 存过或在温箱中温浴2小时以上的板。涂完后建议空气 晾干(20-30分钟)这样会减少卫星菌落出现。 • 预冷是必要的。
分子生物学实验步骤总结超全
一目的基因的获得细菌基因组DNA的提取(一)仪器1)台式高速离心机2)恒温水浴箱3)枪式移液器4)灭菌的EP管和Tip头(二)试剂1)溶液1:25%蔗糖50mmol/LTris-Hcl,pH8.050mmol/LEDTA,pH8.0500ug/ml溶菌酶(现用现配)100ug/mlRNase2)溶液Ⅱ:100mmol/LTris-Hcl,pH8.01%SDS400ug/ml蛋白酶K3)酚:氯仿:异戊醇(25:24:1)4)氯仿:异戊醇(24:1)5)3mol/LNaAc(pH5.2)6)异丙醇或无水乙醇7)70%乙醇8)TE缓冲液:10mmol/LTris-Hcl,pH8.01mmol/LEDTA,pH8.0(三)实验步骤1)5mL细菌过夜培养,5000rpm离心10分钟,去上清液。
2)将菌体细胞悬于250uL溶液1中,37°C水浴保温过夜。
3)加250uL溶液Ⅱ,55°C水浴保温4小时。
4)加0.9倍体积的酚/氯仿/异戊醇(25:24:1),轻轻混匀,于14000rpm离心10分钟,转移水相至新的Ep管,重复步骤4一次。
5)向转移的水相中加入0,9倍体积的氯仿/异戊醇(24:1),轻轻混匀,于14000rpm离心10分钟,转移水相至新的Ep管,重复步骤5一次。
6)向转移的水相中加入0.1倍体积的3mol/LNaAc和0.7倍体积的异丙醇(或2.5倍体积的无水乙醇),冰上放置30分钟。
7)14000rpm离心20分钟,弃上清,用0.5mL70%乙醇洗涤沉淀一次,弃上清,沉淀于室温干燥。
8)将DNA沉淀溶解于15ulTE缓冲液中,-20°C保存。
9)进行DNA含量和纯度检测,琼脂糖凝胶电泳鉴定。
植物DNA的SDS提取法:(一)试验试剂:1)研磨缓冲液:称取59.63gNaCl,13.25g柠檬酸三钠,37.2gEDTA-Na分别溶解后合并为一,用0.2mol/L的NaOH调至pH7.0,并定容至1000ml。
分子生物学实验 实验步骤 李明
5. 分别将两组反应液各转移到新的1.5ml离心管中,加入200ul的LB培养基,37℃复苏培养1小时。
(四)转化子的培养和筛选
1.取复苏菌液100ul,直接涂布于培养基上
2.转化实验组涂布含有Amp的LB培养基上;空白对照组涂布于含有Amp的LB培养基上和不含Amp的LB培养基上。
实验四 PCR技术及产物鉴定
实验步骤
配置PCR反应液于PCR管中
反应液加毕, 混匀,最后瞬时离心把反应液全部集中于PCR管底部。
2.PCR反应:按以下条件进行PCR反应
3.电泳(鉴定)
反应结束后,取3μl反应液,加入少量Loading buffer混匀,进行1%的琼脂糖凝胶电泳(150V)。(Marker由老师操作)。
3.小心吸去上清,加0.7ml培养基通过吹吸悬浮细胞,然后铺到3.5ml培养皿中(预先加入2ml培养基),摇匀;
4.镜检,观察细胞数量和形态;
5.27℃,2-3天后准备用来提取DNA;
实验二动物细胞DNA的提取
操作步骤
1.细胞的收集
(1)将细胞吹起,转入离心管(细胞数量少的补加100-200ul细胞悬液)。 1500g(4000rpm)离心5min,弃上清。
实验六感受态细胞的制备
1.37℃培养菌体约至OD600为0.3-0.4。
2.在洁净台上,每人取培养液一个1.5ml转入1.5ml离心管中,在冰上冷却10min,于4℃,4000rpm/min离心5min.
3.倒掉上清培养液,用750ul的CaCl2溶液轻轻悬浮细胞,冰浴10min. 于4℃,4000rpm/min离心5min.
分子生物学实验技术
《分子生物学实验技术》实验报告实验一碱裂解法小量提取质粒DNA一、实验原理二、在碱性环境中, 细菌膜破裂释放内容物。
染色体DNA变性分开, 而质粒DNA虽变性但仍处于缠绕状态。
将pH调至中性并有高盐存在及低温的条件下, 染色体DNA.大分子量的RNA和蛋白质在SDS的作用下形成沉淀, 而质粒DNA仍然为可溶状态。
通过离心, 可除去大部分细胞碎片、染色体DNA.RNA及蛋白质, 最后用酚、氯仿抽提纯化得到质粒DNA。
三、操作步骤主要步骤1.细菌的培养2.细菌的收集和裂解3.质粒DNA的分离和纯化4.质粒的鉴定具体操作如下:1.取1.5 ml过夜菌液于EP管中, 12 000 r/min离心1min, 弃去上清液。
2.加100 μl 溶液Ⅰ, 剧烈震荡使菌体悬浮均匀, 室温放置5min 。
3.加200 μl 溶液Ⅱ(现配现用), 轻柔颠倒混匀4~6次, 室温放置5min 。
4.加150μl预冷溶液Ⅲ, 轻柔颠倒混匀4~6次, 冰上放置10分钟。
5. 12 000 r/m离心10 分钟(如果上清液仍然浑浊, 可将上清液移出, 再次离心5分钟)。
6.吸出上清液,加2.5倍体积的无水乙醇,混匀,室温放置10分钟,12 000r/min离心10分钟,沉淀DNA 。
7.弃上清,挥干,所得DNA溶于30μlddH2O中,用于后续试验。
三、实验结果获取极少量白色固体, 主要为DNA, 也不排除有蛋白质等杂质, 但是不影响后续实验的继续进行。
实验二氯化钙法进行质粒转化及筛选一、实验原理细菌处于0 ℃, 在CaCl2低渗溶液中, 菌体细胞膨胀成球形, DNA则与CaCl2形成抗Dnase的羟基-磷酸钙复合物粘附于细胞表面, 经42 ℃短时间热冲击处理, 促进细胞吸收DNA复合物, 在丰富培养基上生长数小时后, 球状细胞复原并分裂增殖, 重组子的基因在被转化的细菌中得到表达, 在选择性培养基平板上, 可选出所需的转化子。
分子生物学实验技术实验操作指南
《分子生物学实验技术实验操作指南》目录1 植物基因序列分析 (3)2 引物设计的原则 (4)3 试剂的配置和灭菌 (5)4 植物基因组DNA的提取 (6)5 DNA的定量 (8)6 DNA的电泳 (9)8 PCR产物的回收 (12)9 PCR产物连接T载体和感受态细胞的转化 (14)10 菌落(液)PCR (17)11 质粒DNA的提取 (18)12 质粒DNA的酶切 (20)1 植物基因序列分析1.基因组DNA(genomic DNA, gDNA):功能基因包括三个基本序列:5’上游区,转录区和3’下游区。
2.5’上游区和3’下游区:转录起始位点上游和转录终止位点下游的序列,主要起到基因表达调控作用。
3.转录区:转录起始位点和转录终止位点之间的序列,经核不均一RNA最终被加工为成熟的mRNA。
4.信使RNA(messenger RNA, mRNA):由DNA的一条链作为模板转录而来的,携带遗传信息的能指导蛋白合成的一类单链核糖核酸。
由编码区、上游的5’非编码区和下游3’非编码区组成,5’端带有7-甲基鸟苷-三磷酸帽子结构,3’端有多腺苷酸尾巴。
5.互补DNA(complementary DNA, cDNA):具有与某m链呈互补的序列的。
6.蛋白质编码区(sequence coding for amino acids in protein, CDS):与蛋白质序列一一对应的DNA序列,且该序列中间不含其它非该蛋白质对应的序列。
只有外显子,不含内含子。
7.5’非翻译区(5’-untranslated region, 5’UTR):mRNA位于基因转录起始位点至编码区翻译起始密码子之间顺序。
8.3’非翻译区(3’-untranslated region, 3’UTR):mRNA位于编码区翻译终止密码子下游一段不被翻译的序列。
2 引物设计的原则1.引物长度:一般为20-25bp。
若产物长度等于或小于500bp,可选用16-18bp的引物;若产物长达5kb,则需要用更长的引物。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1.5μl
65 ℃,5min; 4 ℃,1min; DEPC处理水 14μl 5xRT buffer 5μl dNTP 1.5μl RT酶 1μl
2000rpm离心20sec后,42 ℃,60min逆转录;85 ℃,5min终 止反应;4 ℃,3min,防止二级结构形成。
Taq酶
2.5μl
质粒提取(碱裂解法)
• 原理: • EDTA和表面活性剂存在下,经碱处理溶菌,同时 pH 12.0~12.6的碱性环境使细菌线性大分子染色体 DNA氢键断裂,双链解开变性,而质粒DNA的超 螺旋共价闭合环状的双链并不完全分离,在恢复中 性pH并有高盐浓度存在的条件下,质粒DNA又呈 天然构型,而细菌染色体不能复性,相互交联形成 不溶性网状结构,与不稳定的大分子RNA、蛋白质 -SDS复合物等一起,通过离心沉淀而除去。再经酚、 氯仿抽提方法进一步纯化质粒DNA。
注意事项 • 洁净是最重要的。 • 离心力要注意不要超过2500g,建议1500-2000g 5min。 • 涂板要注意,不要觉得不匀而多次反复,轻轻在平板上 带一下感觉板上大部分地方走过即可,特别在冰箱中保 存过或在温箱中温浴2小时以上的板。涂完后建议空气 晾干(20-30分钟)这样会减少卫星菌落出现。 • 预冷是必要的。
二、质粒转化、平板筛选 • 制备含含100μg/ml氨苄青霉素(Amp)的LB琼脂平板 上 (50 ℃ 时加入Amp )
• 取感受态细菌100μl置于ቤተ መጻሕፍቲ ባይዱ菌EP管中;
• 加入重组质粒DNA10 μl,轻轻旋转混匀,冰上放置 30min,42 ℃热休克90sec(勿超时!),冰浴速冷12min。 • 加入无抗生素的LB培养基400μl,于37 ℃摇床(100150rpm)温和振摇45min,使细菌复苏并表达质粒的抗 性基因。 • 取100μl菌液均匀涂铺于含Amp的LB琼脂平板上 , 37 ℃ 20min后,倒置平板继续培养10-16h(小于20h) • 挑取单菌落置5mlLB培养基中(含Amp) 37 ℃摇床培 养8-12h后,提取质粒,限制酶切鉴定。
一、感受态细菌的制备
1.取出液氮或低温冰箱中贮存的大肠杆菌在LB琼脂平板上划线,37 ℃过夜至长 出单菌落。 2. 挑选琼脂培养平板上的单菌落, 接种到含有3 m1 LB培养基的试管内,37℃振 摇过夜。次日取菌液1 m1接种至含有100 m1 LB培养基的500 m1烧瓶中, 37℃剧烈振摇(200~300 rpm)培养约2~3 h,待OD600值达到0.3~0.4时将烧瓶 取出立即置冰浴10~15 min。 自该步骤起皆需无菌操作。 3. 将细菌转移到一个预冷的1.5 ml灭菌的离心管中。于4℃离心,8000 g×1 min回收细胞。
RT-PCR扩增目的基因
第二步:PCR(总体积50μl) 灭菌双蒸水 31.5μl 10xTaq buffer 5μl Mg+2 4μl dNTP 1μl Sense primer 2μl Antisense primer 2μl
cDNA模板 2μl
2000rpm离心20sec后,94℃,1min;56 ℃,1min; 72℃, 1min ,30cycles; 72 ℃,10min 。
操作步骤
•
• • • •
(收集菌液于Ep管中,10000rpm×30 s,弃上清,用1 ml STE漂洗1~2次,离心弃上 清) 1.细菌沉淀加预冷的溶液I 100 μl重悬菌体,强烈振荡混匀。 2.加溶液Ⅱ 200μl,快速颠倒4~6次混匀内容物,冰上3~5 min(勿超时)。 3.加溶液Ⅲ 150 μl,反复颠倒数次混匀,冰上5 min 。 4.离心12 000 rpm×5 min,转上清至新Ep管中。 rpm 5.室温下加等体积饱和酚至样品处理液中,缓慢颠倒混匀10min。 6.离心14000 rpm ×5min,取上层水相到新Ep管中(5,6步可不做)。 7.加等体积氯仿/异戊醇(24:1),充分混匀,离心14000 rpm ×5min,取上层 水相到另一管中。 8.加1/10体积的3 mol/L NaAc(pH5.2)和2.5 倍体积的无水乙醇,混匀,置-20℃ 10 min。 8.离心14000 rpm×10 min,弃上清。 9.沉淀用1 ml 70% 预冷的乙醇洗一次,离心12 000 rpm×5 min,弃上清。 10.将Ep管倒置于滤纸上吸净液体,室温下敞口蒸发痕量乙醇10~15 min 或真空 抽吸2 min。 11.加无菌双蒸水40 μl溶解沉淀。 12.加RNase A 1~2 μl(此步省略), 混匀,置37℃水浴30 min后进行限制性内切 酶酶切实验(见实验六十七),或于-20℃保存待用;1%琼脂糖凝胶电泳鉴定提
实验内容
•感受态细菌的制备 •质粒转化、平板筛选 •质粒的扩增、质粒提取(碱裂解法) •质粒DNA的限制性内切酶酶切及电泳鉴定
•真核细胞基因组DNA的分离纯化(蛋白酶K法) •DNA紫外定性、定量分析及电泳鉴定 •真核组织总RNA提取(TRIzol试剂法) •RNA紫外定性、定量分析及甲醛变性琼脂糖凝胶电泳鉴定 •RT-PCR扩增目的基因 mRNA的反转录 PCR及鉴定
• • • •
• •
取质量
质粒DNA的限制性内切酶酶切
• 反应体系如下: 灭菌双蒸水 10×限制酶buffer 质粒DNA BamHІ HindШ 6μl 2μl 10μl 1μl 1μl
总体积20μl,混匀,37℃水浴1-3 h,,用1%琼脂糖凝胶 电泳鉴定结果。
RT-PCR扩增目的基因
第一步:RT(总体积25μl)
4. 弃去上清,将管倒置于滤纸上 1 min。
5. 加 1ml 4℃预冷的0.1 mol/LCaCl2重悬菌体,冰浴30分钟。 6. 4℃离心1 min, 回收菌体。弃去上清,将管倒置于滤纸上1 min。
7. 加 0.1 ml 4 ℃预冷的 0.1 mol/LCaCl2重悬菌体,置4 ℃冰箱过夜。
即可应用于转化。