重组质粒的原核表达
基因工程载体(质粒)(三)

从不同位点整合到寄主菌染色体上,这种细胞称为Hfr细胞
(高频重组细胞)。
F因子是雄性决定因子,F+ 细胞表面可以形成 一种叫做性须(pilus)的结果,它促使F+ 经 性须进入F- 细胞。F- 细胞则变为F+ 细胞。F因 子可以通过接合作用自我转移,也能够带动寄 主染色体一道转移。但F因子的这种整合过程 是可逆的。在一定条件下,Hfr细胞又可重新 变为F+或F-细胞。 基因工程多选用非接合型质粒,主要安全角度 考虑
常用的是抗生素抗性基因的选择标记。主要有: 四环素(Ter),氨苄青霉素(Amp),氯霉 素(Cm),卡那霉素(Km)。新霉素(Neo) 等。如质粒对氨苄青霉素有抗生时,则写成 AmPr,反之则写成AmPs,。带有抗药性质粒 称为R质粒。一个质粒载体通常有二种抗生素 抗性基因
新的质粒还带有下列标记基因,使于筛选重 组体: ①HA ②Luaferase ③GFP ④便于检测的多肽
非接合型质粒的寄主细胞中同时存在一 种接合型质粒,那么它们通常也是可以 被转移的。这种由共存的接合型质粒引 发的非接合型质粒的转移过程,叫质粒 的迁移作用(mobiligation)又叫质粒 的诱动。
带有大肠杆菌素基因的Col质粒和带有抗菌素抗性基因的R 质粒既有属于接合型的,也有属于非接合型的。 如果在非接合型质粒的寄主细胞中同时存在一种接合 型质粒,那么它们通常也是可以被转移的。这种由共存的 接合型质粒引发的非接合型质粒的转移过程,叫质粒的迁 移作用(mobilization)又叫质粒的诱动。ColE1 是一种可以
钝化了一个抗性基因,保留了另一个抗性基因。插入失活型载体,就是将 外源DNA片段插入到会导致选择性基因(Ampr,Tetr等)失活的位点。如
EGFP的原核表达分析

EGFP的原核表达分析首都师范大学生命科学学院100048摘要:利用PCR反应扩增出所需要的EGFP基因,与pTrc His蛋白表达载体重组,转化大肠杆菌BL21,提取质粒,诱导EGFP蛋白表达,进行进一步的分离纯化并用SDS-PAGE和Western-blot 做检测。
这个实验让我们充分熟悉了整个流程。
关键词:绿色荧光蛋白蛋白的诱导表达、分离纯化表达蛋白检测 E.coli材料和方法:1、主要材料及仪器1.1、菌株和质粒:大肠杆菌BL21为本实验室保存;重组筛选质粒pTrcHis购自Invitroge公司。
1.2、培养基:LB培养基(液/固)内含氨苄青霉素浓度为100 ug/ml。
1.3、器材和试剂器材:移液枪、PCR仪、离心机—(HetticZENTRIFUGEND-78532 Tuttlingen)、SIGMA2-16K(4℃离心机)、SIGMA 3-18K(离50ml大离心管)、紫外透射观察仪、水浴锅、核酸电泳仪(BIO-RAD)、超声波破碎仪(SONICSVIbra cell)、气浴恒温振荡器(THZ-82江苏金坛市金城国胜实验仪器厂)、SDS-PAGE电泳装置、WesternBlotting电转移装置、水平摇床(WD-9405)、胶片观察灯(WD-9406)、封口机、超净台、250ml锥形瓶。
试剂:PCR反应体系缓冲液(10~50 mMTris-HCl,50 mM KCl,1.5 mMMgCl2)、DNA聚合酶(Taq E)、dNTPs混合物(每种2.5mM)、博大泰克PCR产物快速回收试剂盒、限制性核酸内切酶(BglII、EcoRI)、0.5M EDTA、琼脂糖凝胶(1g琼脂糖、100ml1xTAE、10ulEB)、电泳缓冲液(50 xTAE、Tris—乙酸、EDTA)、溴酚兰、蔗糖、甘油、连接酶(购于Promega公司的T4DNA连接酶及10×ligation buffer)、0.1MCaCl2、碱裂解液(溶液I:50mM葡萄糖、25mM Tris-HCl,pH=8.0、10mM EDTA;溶液II:0.2 M NaOH,1%SDS;溶液III:5 M KAc,11.5 ml冰醋酸,加水定容至100ml)、RNaseA、无水乙醇、PBS缓冲液、裂解缓冲液(含20mM咪唑)、冲洗液(含100mM咪唑)、洗脱液(含500mM咪唑)、5×样品缓冲液、30%凝胶贮液、4×分离胶缓冲液、4×浓缩胶缓冲液(4℃保存)、TEMED原液、10%过硫酸铵、Tris-甘氨酸电极缓冲液(pH=8.3)、考马斯亮蓝G250染色液、GFP抗体(200ug/ml,用pH=7.5 TBS配置)、转移缓冲液、TBS、封闭液(5%脱脂奶粉)、显色液(氯萘酚,30mg/ml于甲醇中)、ddH2O、脱色液((100 ml冰乙酸:100 ml乙醇:800 ml蒸馏水)2、方法2.1、EGFP基因的PCR扩增及其电泳检测在一灭菌的0.2mL小离心管中依次加入(总体积为50ml):10×buffer(Mg2+)5ml4×dNTPs (每种 2.5mM )8mlEGFP-F1mlEGFP-R1mlTemplate(模板)34mlTaq E1ml--------------------------------------------------------------------------------Total volume 50µl循环参数94 ℃3min94 ℃30s55 ℃30s 27cycle72 ℃1min72 ℃10min4 ℃保存2.2、PCR扩增产物的回收———试剂盒法1)、柱平衡:向吸附柱CB2中(吸附柱放入收集管中)加入500ml的平衡液BL,12000rpm离心1分钟,倒掉收集管中的废液,将吸附柱CB2重新放回收集管中。
铜绿假单胞菌外膜蛋白OprH原核表达载体的构建与表达

铜绿假单胞菌外膜蛋白OprH原核表达载体的构建与表达罗晓庆;孙济宇;王慧;王晓波;王琪;蒋瑞雪【摘要】目的:克隆铜绿假单胞菌外膜蛋白OprH基因,构建其原核表达载体并鉴定其表达。
方法从铜绿假单胞菌中提取基因组DNA进行PCR扩增OprH基因;采用T-A克隆技术构建pMD19T-OprH重组质粒,并经酶切和序列测定后,获得OprH片段插入到原核表达载体pET28b中,以构建pET28b-OprH重组表达质粒,并在表达宿主菌E�Coli BL21中经IPTG诱导表达及通过SDS-PAGE电泳鉴定。
结果 pET28b-OprH原核表达载体构建成功;重组表达质粒经IPTG诱导后表达外膜蛋白OprH。
结论成功克隆了OprH基因并获得原核表达物,为进一步研究快速检测该菌的方法奠定了基础。
%Objective To clone the gene of outer membrane protein OprH of Pseudomonas aeruginosa ( PA) , construct a prokaryotic expression vector and identify its expression. Method The DNA genome was extracted from PA, the OprH gene was amplified by primersof PCR. The recombinant plasmid pMD19T-OprH was constructed by using T-A cloning. After enzyme digestion and sequence analysis, the OprH gene was inserted into prokaryotic expression vector pET28b to construct recombinant expression plasmid pET28b-oprH, which was expressed in E. coli BL21 (DE3) cells with induction by IPTG. Then the expressed protein was analyzed by SDS-PAGE. Result Construction of the prokaryotic ex-pression vector pET28b-oprH was successful. Then the recombinant expression plasmid expressed a corresponding protein OprH after being induced by IPTG. Conclusion OprH gene was successfully cloned and theprokaryotic expression product was obtained. It provides the basis for investigating a method of rapid detection of PA.【期刊名称】《中国烧伤创疡杂志》【年(卷),期】2014(000)003【总页数】4页(P195-198)【关键词】铜绿假单胞菌;外膜蛋白;原核表达载体;重组质粒【作者】罗晓庆;孙济宇;王慧;王晓波;王琪;蒋瑞雪【作者单位】161006 黑龙江齐齐哈尔,齐齐哈尔医学院免疫教研室;161000 黑龙江齐齐哈尔,齐齐哈尔医学院附属第三医院烧伤科;161006 黑龙江齐齐哈尔,齐齐哈尔医学院免疫教研室;161000 黑龙江齐齐哈尔,齐齐哈尔医学院附属第三医院烧伤科;161006 黑龙江齐齐哈尔,齐齐哈尔医学院免疫教研室;161006 黑龙江齐齐哈尔,齐齐哈尔医学院免疫教研室【正文语种】中文铜绿假单胞菌为条件致病菌,是医源性感染的主要病原菌之一,广泛分布于自然界的土壤、水、空气及人体皮肤、肠道、呼吸道内。
人野生型p53基因的克隆及原核表达

人野生型p53基因的克隆及原核表达王继;高瑞娟;袁大巍;王丽【期刊名称】《吉林大学学报(理学版)》【年(卷),期】2009(47)5【摘要】利用RT-PCR方法由人外周静脉血淋巴细胞中获得人p53 cDNA片段,并将其克隆入原核表达载体pQE40中,构建重组质粒pQE40-p53;转化于E.coliM15宿主菌,经IPTG诱导,表达了N端融合6His的p53融合蛋白.利用6His与Ni2+高亲合力结合的性质,经镍柱纯化、透析袋分级透析复性、 Western Blot鉴定,结果表明获得了纯化的6His-p53融合蛋白.【总页数】4页(P1077-1080)【作者】王继;高瑞娟;袁大巍;王丽【作者单位】东北师范大学,遗传与细胞研究所,长春,130024;东北师范大学,遗传与细胞研究所,长春,130024;东北师范大学,遗传与细胞研究所,长春,130024;东北师范大学,遗传与细胞研究所,长春,130024【正文语种】中文【中图分类】Q78【相关文献】1.卵巢癌的p53基因治疗实验研究——人野生型p53基因真核细胞表达质粒的构建 [J], 关婷;崔满华;李守柔PIO增强MRI检测人野生型p53基因转染动脉粥样硬化易损斑块的研究 [J],陈荣红;祁春梅;李东野;蔡文标;武维恒;姚丽娜;张超3.稳定表达野生型p53基因人骨肉瘤细胞株的建立与鉴定 [J], 雷晓晶;韩鹏飞;吕智4.脉冲电场联合野生型p53基因诱导人宫颈癌Hela细胞凋亡 [J], 李好山;熊正爱;周玮;李成祥;姚陈果;孙才新5.尿刊酸修饰的壳聚糖介导野生型p53基因转染对人肺腺癌细胞系H1299增殖和凋亡的影响 [J], 王伟;姚静;周建平;李夷平;陶磊因版权原因,仅展示原文概要,查看原文内容请购买。
原核表达系统

05
原核表达系统的研究进展
研究现状
基因克隆技术
随着基因克隆技术的发展,越来 越多的基因被成功克隆并用于原 核表达系统中,为生物制品的制 备提供了更多选择。
表达载体构建
原核表达系统中的表达载体是关 键因素,目前已经构建了多种高 效表达载体,能够实现外源基因 的高水平表达。
宿主菌选择
宿主菌的选择对原核表达系统的 表达效果至关重要,经过不断筛 选和改良,已成功应用于生产实 践的宿主菌种类不断增加。
通过原核表达系统可以大量制 备蛋白质,用于研究蛋白质之 间的相互作用和复合物组装。
蛋白质工程改造
利用原核表达系统可以对蛋白 质进行体外进化、定向改造等 ,提高蛋白质的特性和功能。
在生物科学研究中的应用
蛋白质组学研究
生物信息学研究
原核表达系统可用于蛋白质组学研究, 大量制备蛋白质并进行分析,揭示蛋 白质的结构和功能。
原核表达系统
目
CONTENCT
录
• 引言 • 原核表达系统的基本原理 • 原核表达系统的应用 • 原核表达系统的优缺点 • 原核表达系统的研究进展 • 结论
01
引言
主题简介
原核表达系统是一种利用原核生物(如细菌)作为宿主细胞进行 目的基因表达的技术。
它具有操作简便、成本低廉、表达量高等优点,广泛应用于基因 工程、蛋白质工程等领域。
翻译过程中,宿主菌的 核糖体识别mRNA上 的起始密码子,开始翻 译目的基因,合成蛋白 质。
通过调控启动子和终止 子等元件,可以控制目 的基因的表达水平和方 向。
03
原核表达系统的应用
在生物制药领域的应用
80%
生产重组蛋白药物
原核表达系统可用于生产重组蛋 白药物,如胰岛素、生长激素等 ,用于治疗各种疾病。
IPTG-诱导表达
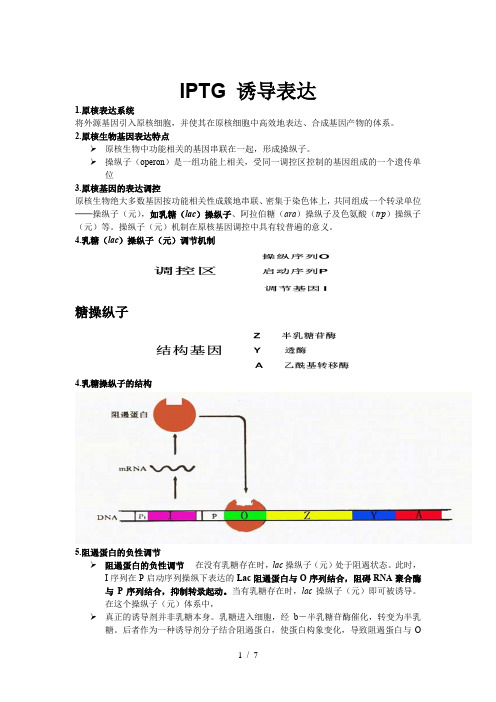
IPTG 诱导表达1.原核表达系统将外源基因引入原核细胞,并使其在原核细胞中高效地表达、合成基因产物的体系。
2.原核生物基因表达特点➢原核生物中功能相关的基因串联在一起,形成操纵子。
➢操纵子(operon)是一组功能上相关,受同一调控区控制的基因组成的一个遗传单位3.原核基因的表达调控原核生物绝大多数基因按功能相关性成簇地串联、密集于染色体上,共同组成一个转录单位──操纵子(元),如乳糖(lac)操纵子、阿拉伯糖(ara)操纵子及色氨酸(trp)操纵子(元)等。
操纵子(元)机制在原核基因调控中具有较普遍的意义。
4.乳糖(lac)操纵子(元)调节机制糖操纵子4.乳糖操纵子的结构5.阻遏蛋白的负性调节➢阻遏蛋白的负性调节在没有乳糖存在时,lac操纵子(元)处于阻遏状态。
此时,I序列在P启动序列操纵下表达的Lac阻遏蛋白与O序列结合,阻碍RNA聚合酶与P序列结合,抑制转录起动。
当有乳糖存在时,lac操纵子(元)即可被诱导。
在这个操纵子(元)体系中,➢真正的诱导剂并非乳糖本身。
乳糖进入细胞,经b-半乳糖苷酶催化,转变为半乳糖。
后者作为一种诱导剂分子结合阻遏蛋白,使蛋白构象变化,导致阻遏蛋白与O序列解离、发生转录。
➢异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定.6.乳糖操纵子的结构及阻遏作用7.外源基因在原核表达系统中表达的必要条件1.删除内含子和5’非编码区2.外源基因置于强启动子和SD顺序控制下3.维持正确开放阅读框架(ORF)4.mRNA稳定且可有效转译,形成的蛋白质不被降解8.影响外源基因在原核细胞中表达效率的因素1、启动子:建立表达载体时,选择强启动子2、基因剂量3、核糖体结合位点9.常见原核强启动子•Plac:受Lac阻遏蛋白负调,受IPTG的诱导•Ptrp:取自大肠杆菌色氨酸操纵子。
•Ptac :Lac启动子和Trp启动子的杂合启动子。
•P L和P R启动子:噬菌体早期左/右向启动子,受λ噬菌体CI基因负调控。
重组蛋白的表达系统(详细版)
• 融合标签: • 融合标签是与目的蛋白共表达的一段多肽,方便重组蛋白的纯化、固定和检测,表3给出了常用的重组
标签。如果不需要对重组蛋白进行纯化,尽量不要引入融合标签,以免影响蛋白性质;如果重组蛋白本 身能够结合某种亲和柱,如某些金属结合蛋白可以结合Ni-NTA,某些糖结合蛋白能够特异识别糖类,也 不必引入标签。 • 融合标签的引入能够大大简化重组蛋白的纯化流程,并提高蛋白溶解度。商业化表达质粒,如pET、 pGEX等提供了各种纯化标签和融合蛋白供选,应根据蛋白具体情况进行选择。 • His-tag是最常用的纯化标签,它具有很多优点:标签较短(10-20个氨基酸残基),不带电(pH8.0), 免疫原性差,通常不影响重组蛋白的结构和功能,Ni2+亲和力高,能够通过一步纯化达到60%-90%的纯 度。 • 如果蛋白质溶解度不高,导致折叠困难、表达量低,可以选择较大的融合标签(GST、MBP、Trx等)帮 助重组蛋白表达和折叠,提高重组蛋白溶解度,从而提高表达量。较大的融合标签有时也会导致翻译困 难甚至提前中止,纯化后发现大部分都是标签蛋白也是常见现象。翻译的提前中止会大大影响重组蛋白 产率和后续纯化,所以在短标签能够达到目的的时候,尽量不要选择大的融合标签。 • 标签位置的选择也很重要:N端标签(短的或长的)自身带有启动子和适应宿主偏好的密码子,可以帮 助目的蛋白表达,提高表达量,但是提前中止翻译的蛋白片段也会被一并纯化出来,降低重组蛋白纯度, 对蛋白酶敏感的、自身容易降解的以及一级序列中有集中的疏水残基区的蛋白尤其要避免使用N端标签; C端标签则可以保证只有完整蛋白得到纯化。另外,如果蛋白的近N端或近C端有重要功能区,如酶活中 心、配体结合位点、二硫键、多聚体稳定界面、相互作用界面等,则要避免纯化标签位于该末端,以免 影响重组蛋白的结构和功能。
植原体免疫主导膜蛋白Imp基因原核表达载体构建及表达
植原体免疫主导膜蛋白Imp基因原核表达载体构建及表达柴化建;赵海泉;张丽君;罗焕亮【摘要】旨在构建植原体免疫主导膜蛋白Imp基因原核表达载体,并进行初步表达.以重组克隆质粒pMD18-T-Imp为模板,PCR扩增Imp基因片段.构建表达载体pET-28a(+)-Imp,转化宿主菌E.coli BL21( DE3).筛选阳性克隆,提取重组质粒作PCR鉴定、酶切鉴定及IPTG诱导表达鉴定.PCR及双酶切结果显示,重组质粒pET-28a(+)-Imp构建成功.经IPTG诱导BL21( pET-28a(+)-Imp)表达约20kD的蛋白,与预期的携带6×His-Tag的目的蛋白(19.5 kD)大小相符,主要以包涵体形式存在.结果显示,构建的表达载体pET-28a(+)-Imp在E.coli BL21( DE3)中能够达一定量表达,为进一步纯化Imp蛋白奠定基础.【期刊名称】《生物技术通报》【年(卷),期】2012(000)006【总页数】5页(P106-110)【关键词】植原体;免疫主导膜蛋白;原核表达【作者】柴化建;赵海泉;张丽君;罗焕亮【作者单位】安徽农业大学生命科学学院,合肥230036;安徽农业大学生命科学学院,合肥230036;深圳市职业技术学院,深圳518055;深圳出入境检验检疫局,深圳518045【正文语种】中文植原体(phytoplasma)是一种存在于植物韧皮部筛管细胞中类似植物病原细菌的单细胞原核生物[1]。
植原体对植物危害严重,可引起植物黄化、小叶、皱叶及丛枝等,因其侵染性强,危害寄主广泛,目前尚无根治方法,易造成严重的经济损失[2]。
由于植原体不能够体外培养,难以获得大量高纯度的植原体,制约了植原体检测防控、致病机制及流行性等研究[3]。
因致病植原体无细胞壁,所以植原体可能通过分泌膜蛋白直接接触寄主而致病。
以此推测植原体膜蛋白在寄主与植原体相互作用过程中起着重要的作用。
PAP蛋白重组表达载体的构建及在原核和真核细胞中的表达
PAP的内化,提高其抗病毒效果。
由于编码PTD的10个氨基酸的核苷酸序列仅为30bp,本实验采用在引物中直接引入该编码序列的方法实现PTD与PAP的基因重组。
同时引入在两个引物(P1和IC。
2)中分别构建原核大肠杆菌表达中所需的BamHI和PstI酶切位点。
具体步骤详见图1。
PTD-prime吖CG曼昼盘里£9_岫HDATGAGGAAGAAGCGGAGACAGCGACGAAGAGTGAATCCAATeATCTACAATG)PTnPAP图1.PTD-PAP的合成模式图通过PCR扩增,用TaqDNApolymerase加A后电泳得到了约800bp的扩增片段。
回收后的片段与pPGEM-TVector连接。
转入大肠杆菌DH5Q中,经蓝白筛选,选取白色菌落行PCR鉴定见可扩增出约800bp的片段;提取PCR阳性菌株的质粒,经BamHI/PstI双酶切,电泳示可约800bp和3Kb处两条条带(见图2)。
株的质粒,经BamHI/PstI双酶切,电泳示可约1Kb和3Kb处两条条带(图4)。
图4pPGEM—TVector/TAT-PAP重组体酶切图示:1.分子量标准为BL2000:2.酶切片段。
图中可见酶切下略大于1Kb片段,相当于Tat加上PAP序列的大小,支持二者重组成功。
将酶切鉴定阳性菌株质粒测序示rI钒序列和PAP序列正确连入pPGEM.TVector中。
1.3.2Tat与PAP的基因重组体中Tat显性负突变体的构建前期的工作共获得的HIV的Tat显性负突变体共六个:pMl:Cys22+Gly;pM2:His33/Cys34一Ala/Ser;pM3:Thr40/Lys41一Al矶~la;pM4:Lys50一STOP;pM5:Ar952一Leu;pM6:Thr40/Lys41/Cys50一Ala/Ala/STOP。
之后验证了突变Tat对HIV-1Tat活性的影响:结果说明不同Tat突变蛋白都对正常Tat有一定程度的抑制作用,其中以pM5最大,达85%,另外pM3可达40%。
质粒表达方向
质粒表达方向质粒表达是一种常用的基因工程技术,用于将目标基因导入到宿主细胞中,使其表达出目标蛋白。
这项技术在基础研究、医学、农业等领域都有广泛的应用。
本文将以质粒表达方向为主题,介绍质粒表达的原理、方法和应用。
一、质粒表达的原理质粒是一种环状的DNA分子,通常存在于细菌细胞中。
质粒表达是通过将目标基因插入到质粒的某个特定位点上,使其与质粒的调控元件相连,从而实现目标基因的表达。
质粒表达的原理可以简化为以下几个步骤:1.构建质粒:选择合适的质粒载体,将目标基因插入到载体的多克隆位点上,并通过限制性内切酶酶切和连接酶连接等技术将目标基因与质粒连接。
2.转化宿主细胞:将构建好的质粒转化到宿主细胞中,使其进入细胞质。
3.选择转化子:通过添加适当的抗生素或其他筛选条件,筛选出已成功转化的细胞。
4.培养和表达:选取转化子,进行培养和表达,使目标基因得以表达。
二、质粒表达的方法质粒表达有多种方法,常用的有原核表达和真核表达两种。
1.原核表达:原核细胞(如大肠杆菌)具有简单的表达系统,是质粒表达的常用宿主。
原核表达的步骤主要包括质粒构建、转化宿主细胞、培养和表达等。
原核表达的优点是操作简单、高效快速,适用于大规模表达和蛋白纯化。
但也存在一些问题,如蛋白质折叠、修饰和活性等方面与真核细胞存在差异。
2.真核表达:真核细胞(如哺乳动物细胞)具有复杂的表达系统,可以更好地保留目标蛋白的结构和功能。
真核表达的步骤包括质粒构建、转染宿主细胞、筛选稳定细胞株、培养和表达等。
真核表达的优点是能够实现蛋白正确的修饰、折叠和定位,适用于研究蛋白的功能和机制。
三、质粒表达的应用质粒表达技术在多个领域都有广泛的应用。
1.基础研究:质粒表达可以用于研究基因的功能、调控和相互作用等。
通过表达外源基因,可以观察其在细胞中的表达情况、蛋白互作和信号传导等过程。
2.药物研发:质粒表达可以用于生产重组蛋白,如抗体、酶和激素等。
这些重组蛋白可以用于药物的生产和治疗。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实验九 表达质粒的构建与诱导表达 第一节 原核表达概述
基因工程的研究一方面是获得基因的表达产物,另一方面是研究基因结构与功能的关系,最终都涉及目的基因的表达问题。表达载体(express vector)是指本身携带有宿主细胞基因表达所需的调控序列,能使克隆的基因在宿主细胞内进行转录与翻译的载体。也就是说,克隆载体只是携带外源基因,使其在宿主细胞内扩增;表达载体不仅使外源基因扩增,还使其表达。 表达载体在基因工程中具有十分重要的作用。外源基因在宿主(受体)细胞内是否表达以及表达水平受到许多因素(调控元件)的制约: 1.正确的阅读框架 为了获得正确编码的外源蛋白,外源基因编码区在插入表达质粒中原核基因编码区时,阅读框架应保持一致。阅读框架是由每三个核苷酸为一组连接起来的编码序列,外源基因只有在它与载体DNA的起始密码相吻合时,才算处于正确的阅读框架之中,从而表达融合蛋白,使外源蛋白与宿主蛋白相融合。 2.目的基因有效转录的启动子 启动子(promoter) 是DNA链上一段能与RNA聚合酶结合并能起始mRNA合成的序列, 它是基因表达不可缺少的重要调控序列。原核启动子是由两段彼此分开且又高度保守的核苷酸序列组成的,分别称为 一35区和 一10区。一35区的序列为TTGACA,一10区序列为TATAAT。此两序列之间的最佳间距为17 bp。可与RNA聚合酶结合,并指导该酶在正确的转录部位开始合成mRNA。由于细菌RNA聚合酶不能识别真核基因的启动子,因此原核表达载体必须用原核启动子带动真核基因在原核生物中转录。原核表达载体启动子的转录常常是可以控制的,即一般情况下不转录,而受诱导剂诱导时就能转录,带动外源基因的高效表达。 目前常用的启动子:如大肠杆菌的lac启动子是乳糖操纵子的启动子,受lacⅠ编码的阻遏蛋白调节控制;trp等启动子是色氨酸操纵子启动子,受trp R编码的阻遏蛋白调节控制;tac启动子,由lac启动子的一l0区和trp启动子的一35区融合而成,汇合了lac和trp两者优点,是一个很强的启动子,受lacⅠ编码的阻遏蛋白调节;lacUV 5启动子是经紫外线诱变改造后的lac启动子,该启动子失去CAP和cAMP的正调控,只要有乳糖或IPTG存在时就能够启动转录;噬菌体的λPL、R启动子是λ噬菌体左、右向启动子,是一个温度敏感的阻遏蛋白受温度调控的很强的启动子;T7噬菌体启动子,比大肠杆菌启动子强得多,并且十分专一,只被T7 RNA聚合酶所识别;SV40启动子、多角蛋白启动子以及花椰菜花叶病毒(CaMV)启动子。 3.转录终止子 构建表达载体时,为了稳定载体系统,防止克隆的外源基因干扰表达载体的稳定性,一般要在多克隆位点的下游插入一段很强的核糖体RNA转录终止子(terminator)。否则合成的 mRNA过长,不仅消耗细胞内的底物和能量,而且容易使mRNA形成妨碍翻译的二级结构。原核基因的转录终止序列包括依赖Rho蛋白和不依赖Rho蛋白两种。不依赖Rho的终止信号序列都含发夹结构,末端为一连串T及YRTCTG。 4.mRNA有效翻译的SD序列 在原核生物中,mRNA分子上有两个核糖体结合位点(ribosome binding site,RBS)也调节着mRNA的翻译,一个是起始密码 (AUG或GUG),另一个是位于起始密码上游3~11个核苷酸处的由3~9个核苷酸组成的一段保守序列AGGAGGU,这段序列富含嘌呤核苷酸,称为SD序列。而核糖体和mRNA的结合是由SD序列以及与起始密码子AUG之间的距离决定的。因此,SD序列和它的起始密码子AUG之间的距离是影响外源基因在原核生物中表达量高低的重要因素。SD序列与AUG间隔9个碱基最为合适,其间距离过长或过短都会影响目的基因的表达,有时由于这种距离的影响,蛋白质合成水平会相差2 000倍以上。且SD序列与AUG之间的碱基最好是A或U,一3位最好是A。 5.转录、翻译后的适当修饰和加工 真核生物基因表达的产物往往需要进行适当的修饰加工,但由于大肠杆菌本身的蛋白质翻译后修饰加工系统相当不完善,表达的真核蛋白质不能形成适当的折叠或糖基化修饰。并且不同宿主其修饰系统也不一样,所以选择宿主时应注意。 6.信号序列(signal sequence) 在原核细胞中,合成的蛋白能否分泌到细胞外与mRNA编码区上游的一段信号序列有关。这段编码序列翻译的15~30个氨基酸为蛋白质N端的信号肽,它能携带蛋白质跨膜并分泌到细胞外。脂双层和负电荷的质膜通过和信号肽的疏水相互作用以及电荷的吸引是信号肽能跨膜分泌的主要原因。当信号肽携带后面的蛋白质跨膜分泌后,信号肽即被质膜上的信号肽酶切除得到有功能的成熟蛋白质。 因此根据不同的需要,人们构建了不同的在原核细胞中高效表达的载体,这些载体通常具有以下几个特点:①有一个强的原核启动子及两侧的调控序列;②位于读码框上游的SD序列与起始密码子AUG之间有合适的距离;③在外源基因和启动子之间有正确的阅读框架;④外源基因下游应加入不依赖ρ因子的转录终止区。一般表达载体都具有多克隆位点,对表达载体和外源基因用相同的两种酶双酶切后,再用T4 DNA连接酶把外源片段连入表达载体中,通过酶切、PCR鉴定,如果插入的片段大小和方向与外源基因一致,则成功构建了重组子。
将克隆的基因插入合适载体后导入大肠杆菌用于表达大量蛋白质的方法一般称为原核表达。这种方法在蛋白纯化、定位及功能分析等方面都有应用。大肠杆菌用于表达重组蛋白有以下特点:易于生长和控制;用于细菌培养的材料不及哺乳动物细胞系统的材料昂贵;有各种各样的大肠杆菌菌株及与之匹配的具各种特性的质粒可供选择。但是,在大肠杆菌中表达的蛋白由于缺少修饰和糖基化、磷酸化等翻译后加工,常形成包涵体而影响表达蛋白的生物学活性及构象。
原核表达载体通常为质粒,典型的原核表达载体应具有以下几种元件: (1)选择标记的编码序列; (2)可控转录的启动子; (3)转录调控序列 (转录终止子,核糖体结合位点); (4)一个多种单一限制性内切酶位点序列; (5)宿主体内自主复制的序列
原核表达的一般程序: 1 获得目的基因 2 准备表达载体 3 将目的基因插入表达载体中(测序验证)——表达质粒的构建 4 表达质粒转化相应的宿主菌 5 外源基因的诱导表达(诱导靶蛋白的表达) 6 表达蛋白的分析
原核表达的基本操作步骤举例: 一、试剂准备 1、LB培养基。 2、100mM IPTG(异丙基硫代-β-D-半乳糖苷):2.38g IPTG溶于100ml ddH2O中, 0.22 μm 滤膜抽滤,-20℃保存。 二、操作步骤 (一)获得目的基因 1、通过PCR方法:以含目的基因的克隆质粒为模板,按基因序列设计一对引物(在上游和下游引物分别引入不同的酶切位点),PCR循环获得所需基因片段。 2、通过RT-PCR方法:用TRIzol法从细胞或组织中提取总RNA,以mRNA为模板,逆转录形成cDNA第一链,以逆转录产物为模板进行PCR循环获得产物。 (二)构建重组表达质粒 1、载体酶切:将表达质粒用限制性内切酶(同引物的酶切位点)进行双酶切,酶切产物行琼脂糖电泳后,用胶回收Kit或冻融法回收载体大片段。 2、PCR产物双酶切后回收,在T4 DNA连接酶作用下连接入载体。 (三) 获得含重组表达质粒的表达菌种 1、将连接产物转化大肠杆菌DH5α,根据重组载体的标志(抗Amp或蓝白斑)作筛选,挑取单斑,碱裂解法小量抽提质粒,双酶切初步鉴定。 2、测序验证目的基因的插入方向及阅读框架均正确,进入下步操作。否则应筛选更多克隆,重复亚克隆或亚克隆至不同酶切位点。 3、以此重组质粒DNA转化表达宿主菌的感受态细胞。 (四)诱导表达 1、 挑取含重组质粒的菌体单斑至2 ml LB(含Amp50μg/ml)中37℃过夜培养。 2、按1∶50比例稀释过夜菌,一般将1ml菌加入到含50mlLB培养基的300 ml培养瓶中, 37℃震荡培养至OD600
≌0.4-1.0(最好0.6,大约需3 h)。
3、取部分液体作为未诱导的对照组,余下的加入IPTG诱导剂至终浓度0.4 mM作为实验组,两组继续37℃震荡培养3hr。 4、分别取菌体1ml, 离心12000g×30 S收获沉淀,用100μL 1%SDS重悬,混匀, 70℃10 min。 5、离心12000g×1min,取上清作为样品,可做SDS-PAGE等分析。 三 、注意事项 1、选择表达载体时,要根据所表达蛋白的最终应用考虑。如为方便纯化,可选择融合表达; 如为获得天然蛋白,可选择非融合表达。 2、在构建表达质粒时,要注意使外源基因与载体DNA的起始密码相吻合时,使其处于正确的阅读框架之中。 3、融合表达时在选择外源DNA同载体分子连接反应时,对转录和转译过程中密码结构的阅读不能发生干扰。 四、评议 1.表达质粒的构建的实验操作基本上与扩增质粒的构建相同,只是构建表达质粒时为了高 效表达外源基因,一般目的基因应与载体的相关DNA片段相融合。融合蛋白在大肠杆菌中的表达的优点如下:融合蛋白N端由正常的大肠杆菌本身序列所控制。容易获得高效表达;融合蛋白往往不被大肠杆菌视为异己蛋白,更为稳定。 2.表达载体与目标基因在体外重组完成后,必须导人特定的受体细胞如:BL21(DE3),使之无性繁殖,直接改变其遗传性状。 3.在外源基因克隆到表达载体时,应注意其序列的ORF的正确与否,它直接决定其编码 的氨基酸序列,并且直接影响蛋白质的功能。如果ORF内碱基对发生缺失突变或插入突变,即插入或缺失非3的倍数的碱基对,那么ORF就可能发生改变,而得到一个截然不同的蛋白质产物,这种变化就是移码突变。如果发生突变形成了终止密码子,就会产生提前终止的、不完整的蛋白质,即无意义突变。其可分三种情况:琥珀突变(amber),原来的密码子突变为UAG;赭石突变(ochre),原来的密码子突变为UAA;乳白石突变(opal),原来的密码子突变为UGA。 4.为了使外源基因在原核细胞中高效表达应利用蛋白酶缺陷型的宿主,如用lon营养缺陷 型减少外源蛋白的降解。因为黄嘌呤核苷(1on)是大肠杆菌合成蛋白酶的主要底物。lon营养缺陷型宿主不能合成黄嘌呤核苷,从而不能合成蛋白酶。
第二节 外源基因的诱导表达 一、实验目的 了解诱导外源基因表达的基本原理,学习和掌握诱导外源基因表达的常用的方法,为SDS-PAGE分析做准备。 二、实验原理 外源基因在原核生物中高效表达除了有适合的载体外,还必须有适合的宿主菌以及一定的诱导因素。宿主菌的选择对外源基因的表达是至关重要的,因为外源基因(特别是真核基因)在细菌中的表达往往不够稳定,常常被细菌中的蛋白酶降解。因此有必要对细菌菌株进行改造,使其蛋白酶的合成受阻,从而使表达的蛋白得到保护。实验中常用的宿主菌是经过改造的JMl09、BL21等大肠杆菌菌株。