CRISPR基因编辑技术教程
基因编辑技术CRISPR

基因编辑技术CRISPR基因编辑技术CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)是一种革命性的基因工程技术,它可以精准地修改生物体的基因组,为科学研究、医学治疗和农业发展带来了巨大的希望和机遇。
CRISPR技术的出现改变了基因编辑领域的格局,被誉为“基因剪刀”,具有高效、精准、便捷等特点,受到广泛关注和应用。
一、CRISPR技术原理CRISPR技术是受到细菌天然免疫系统的启发而发展起来的一种基因编辑技术。
细菌通过CRISPR/Cas系统来抵御病毒入侵,其中CRISPR是一段DNA序列,记录了细菌曾经感染过的病毒基因信息,Cas蛋白则能识别并切割这些病毒基因。
科学家们发现,通过改造CRISPR/Cas系统,可以实现对生物体基因组的精准编辑。
CRISPR技术的基本原理是利用一种叫做“引导RNA”的分子,它能够将CRISPR/Cas系统导向到特定的基因组位置,然后Cas蛋白就会在这个位置上进行切割或编辑操作。
通过设计合适的引导RNA序列,科学家可以实现对基因组的精准编辑,包括基因敲除、基因插入、基因修饰等操作。
二、CRISPR技术在科学研究中的应用CRISPR技术在科学研究领域发挥着重要作用,它为科学家们提供了一种高效、精准的基因编辑工具,帮助他们研究基因功能、疾病机制等重要科学问题。
通过CRISPR技术,科学家们可以快速生成基因敲除或基因突变的细胞系或动物模型,从而揭示基因在生物体内的功能和作用机制。
此外,CRISPR技术还被广泛应用于基因组筛选、基因组编辑、疾病模型构建等方面。
科学家们利用CRISPR技术可以精准地编辑细胞的基因组,研究基因与疾病之间的关系,为疾病的治疗和预防提供新的思路和方法。
三、CRISPR技术在医学治疗中的应用CRISPR技术在医学治疗领域具有巨大的潜力,可以用于治疗各种遗传性疾病、癌症、传染病等疾病。
基因编辑的步骤与流程

基因编辑的步骤与流程基因编辑是一项革命性的生物技术,通过改变生物体细胞内的基因序列,可以使人类对遗传性疾病和其他基因相关疾病的治疗更加精确和有效。
在过去的几十年里,科学家们开发了许多不同的基因编辑技术,其中最常用的是CRISPR-Cas9。
基因编辑的步骤通常包括序列选择、设计寡核苷酸、合成寡核苷酸、递送寡核苷酸和验证编辑效果。
首先,进行基因编辑之前,科学家需要选择要编辑的目标基因序列。
这个选择通常基于已有的研究和对相关基因与特定疾病之间关联的了解。
通过了解基因序列,科学家可以确定需要进行修饰的位点。
接下来,科学家需要设计特定的寡核苷酸(单链DNA或RNA片段),以便与目标基因序列发生特定的交互作用。
这些寡核苷酸通常由20到30个核苷酸组成,拥有与目标基因序列相匹配的部分。
然后,设计好的寡核苷酸序列将被合成出来。
科学家通常将这项任务交给专门的合成公司来完成。
这些寡核苷酸可以通过一系列化学反应合成,并且可定制长度、序列和纯度。
接下来,寡核苷酸需要被递送到特定的细胞中。
通常使用载体系统来实现这一目标,常见的载体系统包括病毒载体和非病毒载体。
病毒载体可以有效地将寡核苷酸递送到细胞内,并确保其被准确地插入到细胞的基因组中。
最后,科学家需要验证基因编辑的效果。
这可以通过PCR扩增和测序等方法来实现。
PCR扩增可以扩增目标基因序列,并通过与已知正常基因序列进行比较来检测编辑的效果。
测序技术可以精确地确定基因序列的每一个碱基,从而验证编辑的一致性。
值得注意的是,基因编辑是一项复杂的技术,涉及到伦理和安全等重要问题。
在进行基因编辑实验之前,科学家们需要遵循严格的伦理和法律规定,并获得相应的批准和许可。
基因编辑技术的发展为人类带来了前所未有的疾病治疗机会。
然而,这项技术还处于不断发展和改进的过程中,其应用仍然存在许多挑战和限制。
随着科学家们对基因编辑技术的深入研究和理解,相信未来将会有更多潜在治疗和应用方面的突破。
(仅供参考)某公司内部的CRISPR-Cas9操作流程
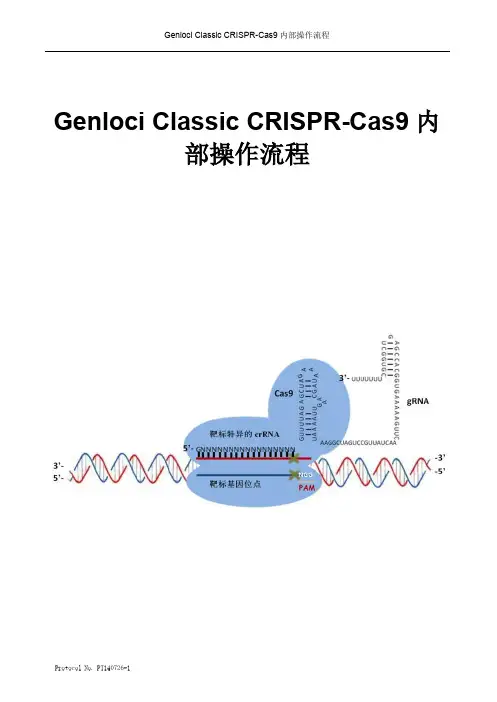
Genloci Classic CRISPR-Cas9内部操作流程Protocol No. PT140726-1Protocol No. PT140726-11,CRISPR-Cas9基因编辑实验流程图如下:pGK1.1U6 CACC G CAAA 4 55’ -5’3’-3’ -- ~20bp2,操作步骤实验前,请您务必做好以下验证实验:A.单细胞生长情况,确保单个细胞可以正常生长形成单克隆,即,低密度的细胞在培养皿中可以形成单克隆。
B.目的基因的表达情况分析,为防止因染色体缺失等情况导致靶基因缺失,首先需要PCR扩增靶基因,并对PCR结果进行测序,确保靶基因的完整存在;其次,您还可以用RT-PCR分析靶基因的活跃度。
1. 设计Oligo DNA序列首先,您需要在靶标DNA区域中设计一对20bp左右的oligo DNA,您可以通过以下在线工具设计:●麻省理工学院的CRISPR Design:/●德国癌症研究中心的E-Crisp:/E-CRISP/designcrispr下面我们选择麻省理工学院的CRISPR Design工具来做设计举例,以Fut8基因为例,一次只能输入大小为23~250bp的基因片段,最好一次只输入一个外显子,避免Guide序列跨内含子的。
点击“Download as genbank”按钮,出现以下界面:“Fut8”Array根据左边的score的高低选取合适的Guide序列,以Guide#1序列为例,2条单链oligo的序列如下(红色字体部分是要与Bbs I酶切后的载体相互补的部分):Fut8-F: cacc G AATGAGCATAATCCAACGCCFut8-R: aaac GGCGTTGGATTATGCTCATTC※注意:oligo DNA设计序列的第一个碱基必须是G,如果你选取的Guide序列的第一个碱基不是G,可自行加一个G上去。
另外,需在位点上下游各设计一条引物,用于后续PCR或测序检测阳性克隆,引物能扩增约300bp 的DNA片段,上游引物距突变位点约100bp,下游引物距突变位点约200bp。
基因编辑技术的原理与实验方法

基因编辑技术的原理与实验方法基因编辑技术是一种能够精确改变生物体基因组的方法,它在医学、农业、生物研究等领域具有重要的应用价值。
本文将重点介绍基因编辑技术的原理和实验方法,以帮助读者了解该技术的基本原理及其实验操作。
一、基因编辑技术的原理基因编辑技术是指通过针对生物体基因组进行特定位点的改变,来实现对目标基因的修饰。
目前最常用的基因编辑工具是CRISPR-Cas9系统。
CRISPR是一种细菌天然免疫系统,它能够识别并切割入侵细菌的外源基因组(如病毒基因组)。
Cas9是CRISPR系统中的一种酶,它作为一个“剪刀”,可以精确切割特定序列的DNA。
基因编辑的主要步骤如下:1. 选择目标基因:首先确定要编辑的目标基因,并确定编辑的目的,如基因突变、插入或删除等。
2. 设计引导RNA(gRNA):根据目标基因的序列,设计合适的gRNA,可以指导Cas9酶精确识别目标序列。
3. 载体构建:将gRNA和Cas9基因组装到载体中,以便在细胞内表达。
4. 导入细胞:通过转染或病毒载体等方式将构建好的基因编辑复合物导入目标细胞。
5. 基因编辑:在细胞内,Cas9酶与gRNA结合,形成一个复合物。
复合物会识别目标位点,引发DNA双链断裂。
细胞为了修复断裂的DNA链,会启动其自身的修复机制。
二、基因编辑技术的实验方法1. CRISPR-Cas9系统CRISPR-Cas9系统的使用便捷、高效且成本相对较低,因此成为最流行的基因编辑工具。
具体操作步骤如下:(1)设计gRNA:选择目标基因组的特定序列,设计合适的gRNA,以便Cas9酶能够识别和切割。
(2)载体构建:将gRNA和Cas9蛋白基因构建到相应的表达载体中。
(3)细胞培养:培养目标细胞(如细胞系或原代细胞)至适当的生长状态。
(4)转染:通过转染方法(如细胞培养基添加转染试剂、电穿孔等方法),将构建好的CRISPR-Cas9复合物导入目标细胞。
(5)筛选和鉴定:筛选转染细胞并分离单克隆,通过PCR、测序等方法检测基因编辑效果。
CRISPR基因编辑技术教程

01
畜禽品种改良
通过CRISPR技术改良畜禽的生长速度 、肉质品质、繁殖性能等性状,培育优 良品种。
02
03
转基因育种
将CRISPR技术与转基因技术相结合, 实现基因的精确编辑和定向转移,加 速育种进程。
04
CRISPR技术挑战与问题
脱靶效应及安全性问题
脱靶效应
CRISPR技术在基因编辑过程中,有时会 出现非特异性切割,导致基因组其他位 置的突变,即脱靶效应。这可能会引发 不可预测的基因功能变化,甚至产生安 全隐患。
通过测序等方法验证表达载体的 正确性和完整性。
转化细胞系或组织
准备细胞系或组织
选择需要编辑的细胞系或组织,并进行适当的预 处理。
转化方法选择
根据细胞类型和组织特性选择合适的转化方法, 如脂质体转染、电穿孔等。
转化效率检测
检测转化效率,确保足够数量的细胞被成功转化 。
验证基因编辑效果
DNA水平验证
通过改进Cas9蛋白的特异性,降低脱靶率,提高基因编辑 的精确度。
开发新型CRISPR系统
探索除CRISPR-Cas9外的其他CRISPR系统,如CRISPRCas12a、CRISPR-Cas13等,以提高编辑效率和特异性。
结合其他基因编辑技术
将CRISPR技术与其他基因编辑技术(如碱基编辑器、先导 编辑器)相结合,实现更高效、更精确的基因编辑。
提取转化后的细胞DNA,通过 PCR、测序等方法检测目标基
因是否被成功编辑。
RNA水平验证
提取转化后的细胞RNA,通过 RT-PCR等方法检测目标基因的 表达水平是否发生变化。
蛋白质水平验证
提取转化后的细胞蛋白质,通 过Western blot等方法检测目 标蛋白质的表达水平是否发生 变化。
CRISPR-Cas9-基因编辑技术简介ppt课件

Cpf1切口远离识别位点。
Feng Zhang et al. 2015
Cpf1 Is a Single RNA-Guided Endonuclea精se选版of课a件Cpplat ss 2 CRISPR-Cas System
21
7.视频:基因编辑CRISPR-Cas9原理
精选版课件ppt
22
精选版课件ppt
它与foki酶功能类似但是它并不需要形成二聚体才能发挥作用2crisprcas系统结构crispr基因座的表达包括转录和转录后的成熟加工当该噬菌体再次入侵细菌时crispr簇首先转录为长的crrna前体然后逐步加工成小的成熟的crrnacrisprcas系统活性的发挥或外源遗传物质的干扰crrna结合相关的cas蛋白后形成crrnacas蛋白复合体通过碱基互补配对精确地与目标dna相结合随后cas蛋白对目标dna进行断裂和降解3cris
Cpf1系统更简单一些,它只需要一条 RNA。Cpf1酶也比标准SpCas9要小, 使得它更易于传送至细胞和组织内。
Cpf1以一种不同于Cas9的方式切割DNA。 Cas9切割留下“平端”(blunt ends)。
Cpf1复合物生成的两条链切口是偏 移的,在裸露端留下了短悬端 (overhang)。这预计有助于精确 插入。
精选版课件ppt
3
1.CRISPR/Cas系统概述
1.2CRISPR系统获得:
CRISPR/Cas系统是很多细菌和大部分古细菌的天然免疫系统, 通过对入侵的病毒和核酸进行特异性的识别,利用Cas蛋白进行 切割,从而达到对自身的免疫。
精选版课件ppt
4
2.CRISPR/Cas系统结构
2.1CRISPR/Cas 主要由两部分组成:
基因编辑技术的操作步骤和注意事项
基因编辑技术的操作步骤和注意事项基因编辑技术是一种能够直接修改生物体基因组的工具,它能够精确地对基因进行改造、删除或添加,以实现个性化定制的目的。
在不久的将来,基因编辑技术将为医学领域、农业领域以及环境保护等领域带来革命性的变化。
然而,尽管基因编辑技术具有巨大的潜力,但其操作步骤和注意事项仍然需要严格遵守和处理。
操作步骤如下:1. 选择适当的基因编辑工具:目前广泛采用的基因编辑工具包括CRISPR-Cas9系统、TALENs(转录活化性核酸酶打靶编辑酶)以及锌指核酸酶(ZFNs)。
根据实验需求和目标基因的特性,选择合适的基因编辑工具。
2. 设计合理的基因编辑寡核苷酸(sgRNA)或引物:根据目标基因序列,设计出具有高特异性和高效率的sgRNA或引物。
确保sgRNA或引物与目标基因序列的配对较好,并避免对其他基因的不必要的影响。
3. 载体构建与转染:将sgRNA或引物插入到合适的载体中,将其转入目标细胞。
选择适当的转染方法,如化学法、电穿孔法或病毒载体法,确保基因编辑工具的高效转染。
4. 检测基因编辑效率:利用PCR扩增、DNA序列分析或者检测基因表达等方法,验证基因编辑工具的编辑效果。
确保编辑效果准确、高效。
5. 选择合适的基因编辑后续处理:根据实验需求,选择合适的后续处理方法。
如将编辑细胞克隆化、使其产生稳定的基因改造后代等。
注意事项如下:1. 遵守伦理规范:在进行基因编辑实验时,必须遵守伦理委员会的规定和道德标准。
确保实验过程和目的符合伦理要求,不涉及未经人类同意的基因改造。
2. 安全操作:基因编辑实验需要在适当的实验室条件下进行,遵循相应的安全操作规程。
确保实验人员的人身安全和实验物质的安全。
3. 验证编辑效果:对于编辑后的细胞或生物体,应进行确认实验,验证编辑效果是否达到预期。
通过相关技术手段进行验证,确保编辑结果的准确性和可重复性。
4. 避免非特异修饰:在进行基因编辑时,必须注意避免对非目标基因的不必要修改。
基因编辑技术的操作指南
基因编辑技术的操作指南基因编辑技术是一种能够直接修改生物体基因组的强大工具,它已经被广泛应用于基础研究、治疗疾病和农业改良等领域。
准确、高效的操作是实现基因编辑技术应用的关键,下面将为您介绍基因编辑技术的操作指南。
1. 选择合适的基因编辑技术基因编辑技术主要包括锌指核酸酶(ZFN)、转录活化因子(TALEN)、CRISPR-Cas9等。
在选择合适的基因编辑技术时,需要考虑编辑效率、特异性和操作难度等因素。
近年来,CRISPR-Cas9技术因其操作简便、高效和广泛适用性而成为研究人员首选的基因编辑技术。
2. 设计合理的核酸序列无论使用哪种基因编辑技术,都需要合理设计核酸序列来实现基因组的精确编辑。
对于CRISPR-Cas9技术,设计sgRNA序列是非常重要的一步。
sgRNA是具有与目标基因组相互作用的RNA分子,它会指导Cas9蛋白靶向到目标基因组,并介导DNA双链切割。
在设计sgRNA序列时,需要选择靶向位点,通常选择20个核苷酸(nt)的序列作为目标。
这个序列应根据生物的基因组信息进行设计,可以使用一些在线工具或软件来辅助完成。
3. 构建基因编辑载体基因编辑载体是将基因编辑工具(如Cas9蛋白和sgRNA序列)引入目标细胞的载体。
对于CRISPR-Cas9技术,常用的载体是质粒。
构建基因编辑载体时,需要合理选择启动子、标签、选择性标记等元素,以确保载体在目标细胞中的高效表达和定位。
构建基因编辑载体的方法有多种,可以通过重组DNA技术、化学合成或基因克隆等方式进行。
4. 选择适当的细胞在进行基因编辑实验前,需要选择适当的细胞来进行操作。
不同的细胞类型可能对基因编辑技术的敏感性、编辑效率以及细胞分裂能力等有所差异。
在选择细胞时,需要考虑研究目的和实验条件,并确保细胞的健康状态和适应性。
5. 实施基因编辑操作经过前期准备工作后,可以开始实施基因编辑操作。
具体步骤包括:5.1 导入基因编辑载体将构建好的基因编辑载体导入目标细胞中。
基因编辑技术CRISPRCas9的应用方法总结
基因编辑技术CRISPRCas9的应用方法总结CRISPR-Cas9基因编辑技术是一种革命性的生物工程工具,它利用细菌体内天然存在的免疫系统来精确修改基因。
自从2012年首次引入以来,CRISPR-Cas9已经被广泛用于改变生物学研究和医学治疗的方式。
本文将介绍CRISPR-Cas9的原理、应用方法,并总结其在不同领域的应用。
### CRISPR-Cas9的原理CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)是一种存在于细菌和古生菌基因组中的DNA序列,它记录了它们所受到的外来病毒基因组的片段。
Cas9是CRISPR系统中的一种蛋白质,具有剪切DNA 的能力。
利用这种系统,科学家们成功将CRISPR-Cas9技术应用于编辑生物体的基因。
CRISPR-Cas9系统的工作原理如下:1. 选择目标基因:确定需要编辑的特定基因序列,并设计与其互补的RNA引导分子(sgRNA)。
2. sgRNA的结合:通过合成互补基因组DNA片段成为单链RNA,与Cas9蛋白结合成一个复合物。
3. 定位到基因组:CRISPR-Cas9复合物进入靶细胞,通过与目标基因的序列互补对应,定位到特定的基因位点。
4. 剪切DNA:Cas9蛋白通过剪切DNA的方式精确修改目标基因,形成双链断裂。
5. 修复机制介入:细胞自身的DNA修复机制介入,通过非同源末端连接(NHEJ)或同源重组(HDR)方式修复双链断裂。
6. 基因修复:通过修复机制,引入目标基因的缺陷或修复,实现基因编辑。
### CRISPR-Cas9的应用方法CRISPR-Cas9技术的应用方法主要包括基因敲除、基因敲入和基因打靶等。
下面将详细介绍这些方法:#### 1. 基因敲除基因敲除是指通过CRISPR-Cas9技术使目标基因完全失活。
其步骤如下:- 设计sgRNA:选择目标基因的外显子序列,设计与之相互配对的sgRNA。
crisprcas基因编辑技术原理与应用 ppt课件
模式
动物 关的基因来用小鼠或其他模型动物建立
相应的疾病模型。研究者可以应用这些
疾病模型研究疾病的发病机制,再现疾病
发生过程;也可用于筛选有效治疗药物,
或通过研究特异的表面分子、信号通路
等开发新的靶向治疗药物等。
17
传统方式
用特定的手段将目的DNA片段插入到小鼠胚胎干细 胞中,筛选出成功修饰的胚胎干细胞,应用显微操 作技术将其移入到早期胚胎(即囊庇)中,将改造后 的脏胎移植到假孕母鼠中,再自生出的小鼠筛选疾 病模型。
利用CRISPRCas进行基因组工程,图中不同颜色的剪刀
代表着不同的Cas9酶
13
应用
用CRISPR/Cas技术绕过了胚胎干细胞操作过程,可快速而有效地建立 携带多个基因突变的小鼠。因其不依赖在胚胎干细胞上操作,可用于多生物 细胞的基因操作。模型生物的建立也不再局限于小鼠及少数大鼠中,任何可 进行胚胎操作的物种都能成为基因组工程的目标。
好 相比其它编辑技术CRISPR/Cas基因编辑技术好用吗?
怎
怎么用CRISPR/Cas基因编辑技术?
7
目录
CRISPR/Cas基因编辑技术
发现 原理 应用 优点 缺点 发展
前景
8
发现
在1987年的一篇论文中,日本大阪大学的研究人员报告了一项表面上微不 足道的研究发现。他们在调查一种编码碱性磷酸酶的细菌基因序列时,发现 了邻近的一个不同寻常的DNA片段,这一DNA片段中间是一段短直接重复 核苷酸序列,两侧为短特异片段。他们当时指出“这些序列的生物学意义尚 不清楚”。在几乎过去快30年后,这一最初看起来不起眼的研究发现,现在 为简易操控大量生物体的基因组打开了大门。2000年,相似的重复序列在其 它真细菌和古细菌中被发现并被命名为短间隔重复序列(Short Regularly
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
段的Tale蛋白。
TALEN原理
TALEN的特异性打靶的原理:
一对可以特异性识别靶基因的TALE,定位到需要编辑的基因组区域; 然后非特异性核酸内切酶 FokI 切断双链 DNA 从而造成 DNA 双链断裂( doublestrand break,DSB); 再通过DNA的自我修复,引起碱基的缺失或突变,从而引起基因突变。 FokI只有在形成二聚体的时候才有内切酶活性。
CRISPR/Cas 9的技术应用
近日,中国科学家利用基因编辑技术—— CRISPR/Cas9,对抑制狗骨骼肌生长的基 因(MSTN)进行了敲除,培育出两只肌 肉发达的“大力神”狗,成功构建了世界 首个基因敲除狗模型。
三大基因修饰技术比较
CRISPR/Cas 9系统的优势
操作简单,靶向精确性更高。sgRNA靶向序列和基因组序列 必须完全匹配,Cas9才会对DNA进行剪切。编码sgRNA 的序 列不超过100bp,因此比构建TALENs和ZENs更简单方便, 用于CRISPR的sgRNA识别序列仅需20个核苷酸。 CRISPR/Cas9系统是由RNA调控的对DNA的修饰,其基因修 饰可遗传。 基因修饰率高,CRISPRs基因敲入的效率为51%-79%, TALENs的效率为0%-34%。 基因调控方式多样,例如敲除、插入、抑制、激活等 无物种限制,可实现对靶基因多个位点同时敲除。 实验周期短,最快仅需2个月,节省大量时间和成本。(ZFNS 一个靶点成本6000$)
CRISPR-Cas系统简介
CRISPR-Cas系统的研究历史
1987 年,日本课题组在K12 大肠杆菌的碱性磷酸酶基因附近发现串联间 隔重复序列,随后发现其广泛存在于细菌和古细菌的基因组中。 2002 年, 正式将其命名为成簇的规律间隔的短回文重复序列 2005 年发现CRISPR 的间隔序列(spacer)与宿主菌的染色体外的遗传物质 高度同源,推测细菌可能通过CRISPR 系统可能以类似于真核生物的 RNAi 方式抵抗外源遗传物质的入侵。
应用
基因组巡航导弹,分子剪刀,DNA外科医生 定点突变,定点修饰(包括得到转基因),转录调控 基因治疗(如遗传病)
基因编辑的三大利器
• ZFN (Zinc-finger nucleases, ZFN) • TALEN(transcription activator-like effector nucleases) • CRISPR/Cas (clustered regulatory interspaced short palindromic repeat /Cas-based RNA-guided DNA endonucleases)
核酸内切酶: 非特异性核酸内切酶FokI,形成二聚体时切割双链DNA
TALEN原理
全长为34aa的Tal 蛋白的第12、13位氨基 残基可以特异性识别核苷酸碱基。 • NG可以识别T • HD可以识别C • NI可以识别A • NN可以识别G或A 通过这种特异性识别,将多个Tal蛋白组 装在一起,就可以构成可以识别目的片
TALEN介导的基因编辑
TALEN质粒对共转入细胞后,表达两个融合蛋白,分别与靶位点特异结
合,由于两个TALEN融合蛋白中的Fok I临近,形成二聚体,发挥非特 异性内切酶活性,在两个靶位点之间剪切DNA。
形成DSB时,同源重组可将需要敲入的序列组合入基因组。
TALEN的技术特点
• 任意敲除,无基因序列、细胞、物种限制,永久失活
并引起一系列生物伦理的问题…
基因组编辑技术
• 基因组编辑技术(genome editing)是一种可以在基因 组水平上对DNA序列进行改造的遗传操作技术。也称为基 因打靶(Gene targeting)。 • 技术的原理是构建一个人工内切酶,在预定的基因组位置 切断DNA,切断的DNA在被细胞内的DNA修复系统修复 过程中会产生突变,从而达到定点改造基因组的目的。
两条ZFN之间具有被称为“间隔区”的spacer结构,该结构的长度以5~6bp 为宜,7bp也能正常工作,合理的“间隔区”设计才能保证ZFN二聚体拥有最 佳的工作空间。
9
ZFN技术的缺陷
• DNA识别域虽然具有较强的特异性识别能力,但是其识别的序列长度 比较有限。
• 因为ZFN剪切的过程不完全依赖同源二聚体的形成,所以一旦形成异 源二 1993年前ZFN就已开始出现,迄今已发展了20多年才有今天的成就; 2009年底出现的TALEN似乎一夜之间呈现出取代ZFN的架势; 2012年底CRISPR/Cas已显现出取代TALEN的巨大潜力,在2013年成为现实。
荣誉
1. 2011年:ZFN, TALEN和Meganuclease—2011年度方法(Nature Methods) 2. 2012年:TALEN — 2012年十大突破(Science) 3. 2013年:CRISPR/Cas被认为是诺贝尔奖级别的成果,华人科学家张峰已 因此获得生物医学大奖。
现
基因突变技术:物理诱变、化学诱变… 转基因技术:T-DNA插入
状
1. 无法控制突变位点 2. 不能精确控制外源基因插入位点 3. 对靶基因抑制不彻底、不特异 4. 难以稳定的遗传 5. 费时费力费钱,难以大量应用
RNA干扰技术:dsRNA、Artificial miRNA 核外遗传技术:质粒、人工染色体 同源重组技术:e.g. 传统的基因敲除小鼠
• 如果出现脱靶,可能导致DNA的错配和序列改变,产生较强的细胞毒 性。当这些不良影响积累过多,超过细胞修复机制承受的范围时,便 会引起细胞的凋亡。 • 另一方面,该手段在细胞内部操作的精确程度不足,则可能会引起相 关基因突变,引发癌症等。
• ZFN 作为基因治疗的手段之一,如果在生物体内使用,可能会引发免 疫反应。而现有的研究手段尚不能预测引入的ZNF蛋白是否会引起免 疫系统的进攻。
特异性 DNA识别域
+
非特异性 核酸内切酶
ZFN原理
ZFN=蛋白的DNA识别域+核酸内切酶
DNA识别域: 由一系列 Cys2-His2锌指蛋白(zinc-fingers)串联组成(一般3~4个),每 个锌指蛋白识别并结合一个特异的三联体碱基。
8
核酸内切酶: 非特异性核酸内切酶FokI,形成二聚体时切割双链DNA
CRISPR/Cas 9 背景
• 由这些序列和基因组成的系统我们称之为 CRISPR/Cas系统,而在这个系统中常用到 的核酸内切酶为Cas9,所以通常称该系统 CRISPR/Cas9 系统。利用这个系统,细菌 可以不动声色地把病毒基因从自己的染色 体上切除,这是细菌特有的免疫系统。
CRISPR/Cas 9概述
基因组编辑技术
姓名:
学号:
• 自从证明DNA是遗传物质以后,人类就开始了通 过改变DNA来改造生物体生理机理和功能的尝试。
1973年,斯坦利·N·科恩(Stanley N. Cohen)和赫伯特·W·博耶 (Herbert W. Boyer) 成功将蛙的DNA插入到细菌中。 20世纪70年代,基因泰克(Genetech)公司对大肠杆菌进行基因改造, 使其带有一个人源基因(这个基因是人工合成的),最后生产出治疗糖尿 病的胰岛素。 1974年,,加利福尼亚州索克生物研究所(Salk Institute for Biological Studies)的Rudolf Jaenisch通过将SV40 病毒的DNA注射到小鼠的囊胚中, 培育出了第一只转基因小鼠。
• 成功率高,在保证转染效率的前提下,有效性可达95%以上
• 打靶效率高,可完全替代RNAi技术
• 脱靶率低,尚未发现明显脱靶效应 • 技术简单,只需按照常规构建质粒方法构建Talen质粒
CRISPR/Cas 9 背景
CRISPR是生命进化历史上,细菌和病毒进行斗争 产生的免疫武器,简单说就是病毒能把自己的基因整合 到细菌内,利用细菌的细胞工具为自己的基因复制服务, 细菌为了将病毒的外来入侵基因清除,进化出很多成簇 的、规律间隔的短回文重复序列(既CRISPR序列)和 CRISPR相关基因,这一序列首先由日本学者于1987年 首次发现(1),于2002年被Jansen等将正式命名(。
2007 年,Barrangou 等首次发现细菌可能利用CRSPR 系统抵抗噬菌体入 侵;2008 年,Marraffini 等发现细菌CRISPR 系统能阻止外源质粒的转 移,首次利用实验验证了CRISPR 系统的功能
2013 年初,MIT 的研究组首次利用CRISPR/Cas9 系统对人293T 细胞 EMX1 和PVALB 基因以及小鼠Nero2A 细胞Th 基因实现了定点突变。同 年Mali 利用CRISPR/ Cas9 在人293T 细胞和K652 细胞基因的靶位点形成 双链或单链的切口,从而激活细胞的DNA 修复机制高效介导外源基因定 点插入。
Cas蛋白是一种双链DNA核酸酶,能在 guide RNA引导下对靶位点进行切割。它 与folk酶功能类似,但是它并不需要形 成二聚体才能发挥作用。
CRISPR/Cas系统的基本结构
切割作用
识别作用
CRISPR系统通常包括:由不连续的重复序列(repeats,R) 与长度相似的间区序列(spacers,S)间隔排列而成的CRISPR簇, 前导序列(leader,L)以及一系列的CRISPR相关蛋白基因(cas)。 Cas(CRISPR associated):存在于CRISPR位点附近,是 一种双链DNA核酸酶,能在guide RNA引导下对靶位点进行切 割。它不需要形成二聚体才能发挥作用。
CRISPR-Cas:一种来源是细菌获得性免疫的由RNA指 导Cas蛋白对靶向基因进行修饰的技术。
CRISPR-Cas系统的结构
CRISPR-CAS 系统的组成主要包括: 由不连续的重复序列 R( repeat) 与长度相似的间区序列S( spacers) 间隔排 列而成的CRISPR 簇,前导序列L( leader) 以及一系列 CRISPR 相关蛋白基因cas。