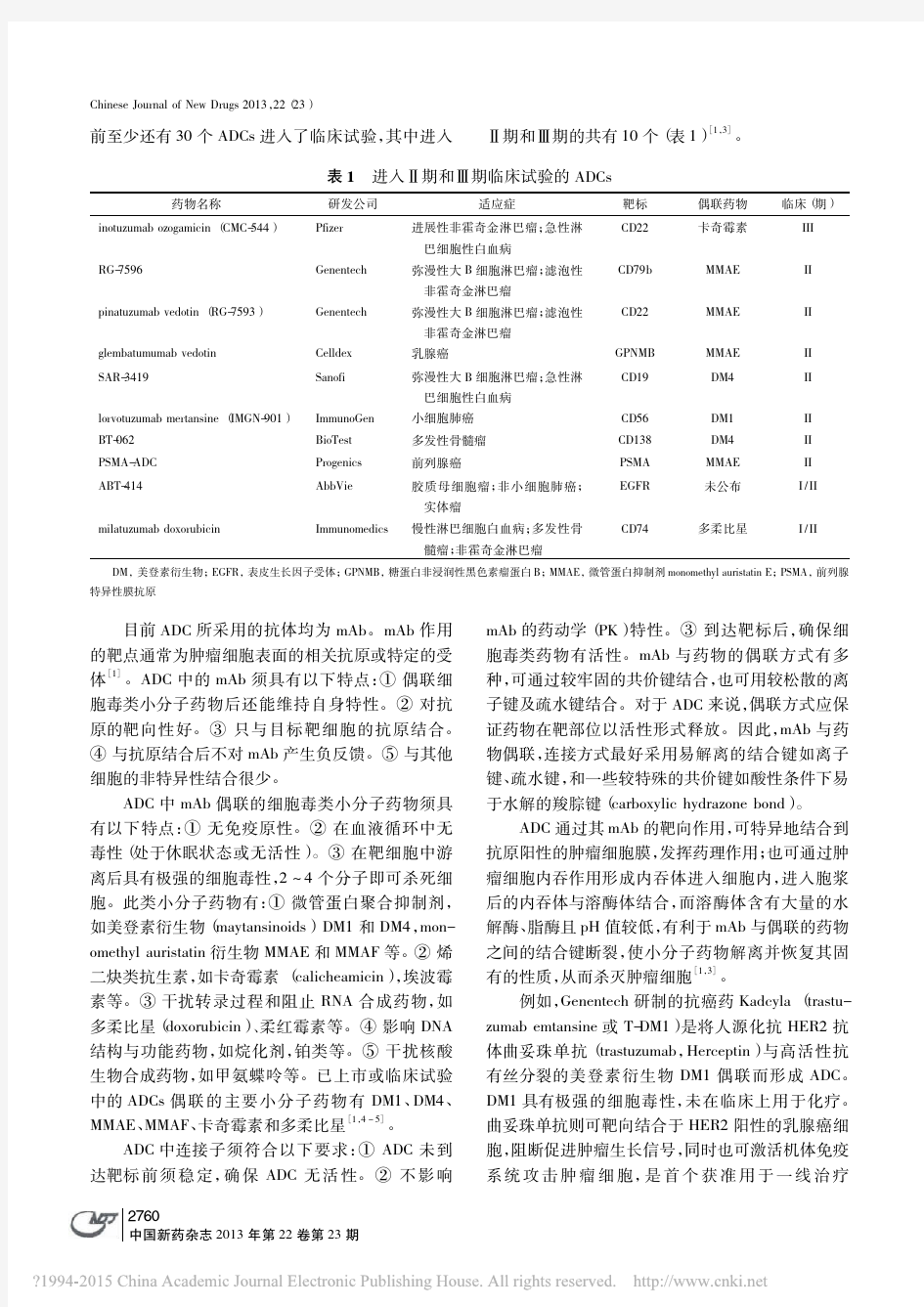
抗体偶联药物临床前安全性评价关注点_宗英

抗体偶联药物中的生物分析
抗体偶联药物研发中的生物分析 李秀立, 陈笑艳, 钟大放* (中国科学院上海药物研究所, 上海药物代谢研究中心, 上海201203) 摘要: 抗体偶联药物(antibody-drug conjugates, ADCs) 是一类单克隆抗体通过一段连接臂共价偶联细胞毒性小分子化合物而成的复合物, 可以提高抗肿瘤药物的靶向性并减少毒副作用。ADCs结构具有异质性并且其药物?抗体比值(drug-to-antibody ratio, DAR) 在体内呈动态变化, 其生物分析面临着巨大的挑战, 常用的定量分析包括酶联免疫吸附反应(ELISA)和液相色谱?质谱分析(LC-MS)。ADCs同其他生物制品一样, 在体内可能会产生抗药抗体(anti-therapeutic antibody, ATA), 影响其药效、药动学及安全性, 因此有必要评价其免疫原性。本文综述了在ADC研发过程中常见的基于ELISA和LC-MS方法的待测物分析, 包括DAR分布、总抗体、结合型抗体、结合型药物、游离药物以及ATA分析, 可为我国的ADCs研发提供参考。 关键词: 抗体偶联药物; 药物?抗体比值; 免疫原性; 酶联免疫吸附反应; 液相色谱?串联质谱 中图分类号: R917 文献标识码:A 文章编号: 0513-4870 (2016) 04-0517-12 Bioanalysis in the development of antibody-drug conjugates LI Xiu-li, CHEN Xiao-yan, ZHONG Da-fang* (Shanghai Center for Drug Metabolism and Pharmacokinetics Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China) Abstract: Antibody-drug conjugates (ADCs) are complex molecules with cytotoxic small molecular drugs covalently bound to monoclonal antibodies via a linker and can improve the targeted drug delivery with minimizing the systemic toxicity. ADCs are heterogeneous mixtures with different drug-to-antibody ratios (DARs) and the DAR distribution is dynamically changing in vivo, therefore the bioanalysis of the ADCs is challenging. Enzyme-linked immunosorbent assay (ELISA) and LC-MS have been widely used in the ADCs bioanalytical assays. Just like other biotherapeutics, ADCs may elicit the host immune response and produce the anti-therapeutic antibody (ATA), which could affect its efficacy, pharmacokinetics, and safety. It is thereby important to investigate its immunogenicity in the ADC development. In this review, we summarized the ELISA- and LC-MS-based bioanalysis strategies for the development of ADCs, including DAR distribution, the determination of total antibody, conjugated antibody, conjugated drug, free drug, and ATA, with the expectation of providing insights and reference for the ADC development in China. Key words: antibody-drug conjugate; drug-to-antibody ratio; immunogenicity; enzyme-linked immunosorbent assay; LC-MS 抗体偶联药物(antibody-drug conjugates, ADCs) 是一类单克隆抗体通过一段连接臂(linker) 共价偶 收稿日期: 2015-09-08; 修回日期: 2015-10-11. *通讯作者 Tel / Fax: 86-21-50800738, E-mail: dfzhong@https://www.360docs.net/doc/f82963230.html, DOI: 10.16438/j.0513-4870.2015-0792联细胞毒性小分子化合物而成的复合物, 用于治疗恶性肿瘤。单克隆抗体可以靶向结合肿瘤细胞表面的抗原, 通过细胞内化作用进入细胞。进入细胞后, 其结构中的小分子药物在溶酶体低pH环境或蛋白酶作用下释放出来, 发挥细胞毒作用。由于单克隆抗体的靶向性, 正常组织细胞内药物浓度较低。因此与传统
抗体偶联药物
抗体偶联药物(ADC的涅槃重生 抗体偶联药物(antibody-drug conjugate, ADC )是将抗体与细胞毒性药物连接起来,通过抗体的靶向作用将细胞毒药物靶向肿瘤,进而降低化疗中常见的 药物非特异性的全身毒性。抗体偶联药物(antibody-drug conjugate, ADC )的研究可以追溯到1980s,,但是直到2000年,首个抗体偶联药物gemtuzumaboz ogamicin (商品名Mylotarg,Pfizer研发)才被FDA B准用于治疗急性粒细胞白血病,但由于偶联技术、靶向性、有效性等受限,完整的抗体偶联药物在血液不稳定,导致致死性毒性的产生,于2010年撤市。这使得本就不明朗的ADC药物研究,更蒙上了一层阴影。 但是随着Takeda/Seattle Genetics 通过对原有技术的改进,利用自己的 新型抗体偶联技术开发了brentuximabvedotin (SGN-35商品名Adcetris ,) 新型抗体偶联药物,并与2011年被FDA批准用于治疗霍奇金淋巴瘤和系统性间变性大细胞淋巴瘤。2013年抗体偶联药物再次取得突破,Ge nen tech/Immu noGen 联合开发的Ado-trastuzumabemtansine (T-DM1,商品名Kadcyla )被FDA批准用于HER2阳性乳腺癌,这是首个针对实体瘤的抗体偶联药物。随着这两个药物的研发成功,ADC药物再次以火热的状态进入人们的研究视野。 1、进入临床阶段ADC药物 截至目前大概有30多种ADC药物进入临床开发阶段(表1),统计表中30 种药物针对适应症发现,其中仅有4种药物针对实体瘤。主要原因:抗体难于透过毛细管内皮层和穿过肿瘤细胞外间隙到达实体瘤的深部。而使用抗体片段,如Fab,制备分子量较小的偶联物,可能提高对细胞外间隙的穿透性,增加到达深部肿瘤细胞的药物量。因此“抗体的小型化或适度的小型化将会是研制ADC药物 的重要途径”。同时我们还能看到ImmunoGe、Seattle Genetics 在现有ADC 药物研发中占有绝对的统治地位,这得力于他们成熟的抗体偶联技术一一利用天然抗体自身的赖氨酸和半胱氨酸中的巯基偶联药物(non-specific )。 2、如何才能成功开发出一种ADC药物?
上市前药物临床安全性评价与风险评估(五)--临床试验安全性数据的总结与评价
发布日期20071130 栏目化药药物评价>>临床安全性和有效性评价 上市前药物临床安全性评价与风险评估(五)--临床试验安全性数据的总结与评标题 价 作者杨焕 部门 正文内容 审评五部杨焕 摘要:风险评估即患者获益与风险比最大化的管理思路应贯穿于药物 的整个生命周期。任何一个新药的风险评估都涉及到数量和质量两个 方面,数量是指安全性数据库的规模;质量是指临床试验设计、实施、 结果分析全过程的质量,在安全性方面具体是指对于药物不良反应报 告、归类、判断、分析总结的质量。本系列文章从技术评价角度,在 分析了解国内外药物不良反应监测的历史与发展的基础上,对上市前 安全性数据库的规模大小和如何保证安全性数据库的质量方面问题进 行了探讨,同时也参考和介绍了国外发达国家最新的风险评估和风险 控制理念,其目的是为临床试验的研究者和注册申请人提供参考和建 议。
关于临床试验中的安全性数据如何进行总结与评价,自上世纪90年代ICH就相继发布了药品注册的国际技术要求,如ICHE2/E3/E6/E9的内容;国内SFDA在2005年也相继发布了《化学药物临床试验报告的结构与内容技术指导原则》和《化学药物和生物制品临床试验的生物统计学技术指导原则》,建议同时参考相关内容。 一、安全性数据的总结和分析 一般原则上要求,只要使用过至少一次受试药物的受试者均应列入安全性分析集。对安全性数据的分析总结应在三个层面加以考虑。首先,必须确定受试者用药/暴露(exposure)的程度,指试验药物的剂量、用药时间,用药的受试者人数,来决定研究可在多大程度上可以评价安全性;其次,确认常见的不良反应、异常改变的实验室检查指标,通过合理的方法进行分类,以合适的统计方法再比较各组间的差异,通过各治疗组之间比较分析出那些可能影响不良反应/不良事件发生频率的因素,如时间依赖性、与剂量或浓度关系、人口学特征等;最后,通过分析因不良事件(不管是否与药物有关)而退出研究或已死亡的受试者进行分析,来确定严重不良事件和其他重度不良事件(指需要采取临床处理,如停药、减少剂量和其他治疗手段的不良事件)。 所有不良事件应明确与药物的因果关系。建议以图表的方式对出现的不良事件进行总结;对患者个体数据列表;对重点关注的不良事件进行详细地描述。受试药和对照药出现的不良事件均应报告。 1. 用药/暴露的程度
创新医药抗体偶联药物发展分析
广州创亚企业管理顾问有限公司 创新医药抗体偶联药物发展分析
已获FDA批准上市的ADC药物(截至2019.11)抗体偶联药物(Antibody–Drug Conjugates,ADCs)已经成为当前全球抗体药 物研发的热门方向。截至2019.11,FDA共批准 了6款ADC药物上市,包括Seattle的Adcetris、 Genentech的Kadcyla和Polivy、Wyeth的 Besponsa和Mylotarg,其中的代表产品——抗 CD30的Adcetris在2018年实现销售收入4.77 亿美元(+55%),抗HER2的Kadcyla则实现销 售收入9.79亿瑞士法郎(+8%)。 该类药物的优点是能够将抗体的高特异性 与细胞毒药物的高杀伤力相结合,对部分肿瘤 展现出优秀的疗效,具有独特的临床价值。
ADC类药物结构示意图抗体偶联药物的设计思路是将抗体 与细胞毒药物进行偶联,从而同时发挥 抗体高特异性与细胞毒小分子的高毒性, 利用抗体-抗原的高度靶向结合将药物 输送至肿瘤部位,将细胞毒药物强大的 细胞杀伤能力集中于肿瘤细胞,降低正 常组织的毒副作用。
ADC药物作用机制 上世纪60年代便已经有了在动物模型中试 验ADC的尝试,80年代推进了临床,首个获批的 ADC是由Wyeth研发的Mylotarg(Gemtuzumab Ozogamicin),于2000年被FDA批准上市。但是 该药物在上市后的临床研究中联用化疗未能延 长生存期且增加了毒性,因此2010年由企业主 动撤市。Mylotarg上市后的10余年间未再有新 的ADC获批。
干货抗体偶联药物(adc)深度研究报告
干货抗体偶联药物(adc)深度研究报告 目录一、行业背景 针对病症 历史沿革基本原理介绍二、核心技术抗体部分(1)靶点选 择(2)抗体选择(3)抗体修饰(4)抗体内吞连接物部分 (1)连接物(Linker)选择(2)连接方式及DAR(3)新技术Abzena的ThioBridge毒素部分核心专利-连接物与毒素排序三、效果对比:Kadcyla对比Herceptin四、核心公司竞 争情况1.领先公司(1)Seattle Genetics (2)ImmunoGen (3)Immunomedics 2.规模较大公司 (1)Abzena (2)Agensys (3)Celldex (4)Progenics Pharmaceuticals (5)Genmab (6)Sorrento旗下Concortis 3.大型药企在ADC领域的布局(1)Abbvie 及其旗下Stemcentrx (2)Roche旗下Genetech的情况(3)武田旗下Takeda Oncology (4)辉瑞ADC 产品Mylotarg (5) 复星、药明、浙江医药与Ambrx (6)三生制药及三生国健(7)丽珠医药集团旗下丽珠单抗 (8)
江苏恒瑞医药(9)四川恒康旗下上海美雅珂生物 (10)其他药企五、行业市场规模六、ADC成功要素分 析独家技术优秀团队研发方向 一、行业背景抗体偶联药物Antibody-drug Conjugate (ADC)是拥有强细胞毒性的化疗药物通过连接物与单抗偶联形成的,兼具小分子药物强大的杀伤力和纯单抗高度的靶向性,因而成为肿瘤靶向治疗的研究和发展热点。但是,ADC 本身并非在各方面强于纯单抗。其疗效的显著提升是通过牺牲药品的均一性与稳定性实现的。现在比较成熟的两种偶联技术分别侧重均一性与稳定性,有一些新式偶联技术能够在两方面同时改善。1、针对病症ADC药物被用于癌症治疗,其针对病症由其中抗体所针对的靶点决定,能够对将该靶点高表达的肿瘤细胞进行针对性DNA破坏或抑制微管。由于 其针对性很高,其可以使用化疗中不能使用或剂量不能提高的高毒性药物[1]。ADC药物相对于化疗药的治疗安全窗口therapeutic window会更大,相对更加安全[2]。下表为可供使用的各种靶点及其针对的癌症种类。其中,目前已经上市的两款药物中,Kadcyla使用HER2靶点,针对HER2阳性的肺癌;Adcetris使用CD30靶点,针对CD30阳性的霍奇金淋巴瘤Hodgkin Lymphoma (HL)与间变性大细胞淋巴瘤anaplastic large cell lymphoma (ALCL) [3]。靶向药物中不同抗原及其针对癌症种类,以及相应在研的ADC药物数量抗
生物药物安全性评价
生物药物安全性评价 第一节生物类药物概述 一、生物类药物的概念和种类 ?生物类药物(biopharmaceutics或biopharmaceuticals)是利用生物体、生物组织或器官等成分,综合运用生物学、生物化学等学科的原理与方法制得的天然生物活性物质以及人工合成或半合成的天然物质类似物。 ?生物药物主要包括生化药物(biochemical drugs)生物技术药物 (bio-technology drugs)、和生物制品(biological products)等。 1、生化药物:一般是系指从动物、植物及微生物提取的,亦可用生物-化学半合成,或用现代生物技术制得的生命基本物质,如氨基酸、多肽、蛋白质、酶、辅酶、多糖、核苷酸、脂和生物胺等,以及其衍生物、降解物及大分子的结构修饰物等。 2、生物技术药物:是指生物来源的和使用生物工程技术制造的药物,包括多肽、蛋白质及其衍生物或由其组成的产品,如细胞因子、生长因子、单克隆抗体、重组DNA 蛋白疫苗及人组织提取的内源性蛋白等。 3、生物制品:是根据免疫学原理,用微生物(细菌、病毒、立克次氏体以及微生物的毒素等)、动物的血液、组织制成的,用以预防、治疗以及诊断人或动物传染病的一类药品。 包括: ★治疗用生物制品:抗体、DNA重组技术制品等。 ★预防用生物制品:疫苗。 ★诊断用生物制品:各种抗原抗体诊断液等。 (一)治疗用生物制品 1.未在国内外上市销售的生物制品。 2.单克隆抗体。 3.基因治疗、体细胞治疗及其制品。 4.变态反应原制品。 5.由人的、动物的组织或者体液提取的,或者通过发酵制备的具有生物活性的多组份制品。
生物药物安全性评价
-生物药物安全性评价
————————————————————————————————作者:————————————————————————————————日期: ?
生物药物安全性评价 第一节生物类药物概述 一、生物类药物的概念和种类 ?生物类药物(biopharmaceutics或biopharmaceuticals)是利用生物体、生物组织或器官等成分,综合运用生物学、生物化学等学科的原理 与方法制得的天然生物活性物质以及人工合成或半合成的天然物质类似 物。 ?生物药物主要包括生化药物(biochemical drugs)生物技术药物 (bio-technology drugs)、和生物制品(biologicalproducts)等。 1、生化药物:一般是系指从动物、植物及微生物提取的,亦可用生物-化学半合成,或用现代生物技术制得的生命基本物质,如氨基酸、多肽、蛋白质、酶、辅酶、多糖、核苷酸、脂和生物胺等,以及其衍生物、降解物及大分子的结构修饰物等。 2、生物技术药物:是指生物来源的和使用生物工程技术制造的药物,包括多肽、蛋白质及其衍生物或由其组成的产品,如细胞因子、生长因子、单克隆抗体、重组DNA蛋白疫苗及人组织提取的内源性蛋白等。 3、生物制品:是根据免疫学原理,用微生物(细菌、病毒、立克次氏体以及微生物的毒素等)、动物的血液、组织制成的,用以预防、治疗以及诊断人或动物传染病的一类药品。 包括: ★治疗用生物制品:抗体、DNA重组技术制品等。 ★预防用生物制品:疫苗。 ★诊断用生物制品:各种抗原抗体诊断液等。 (一)治疗用生物制品 1.未在国内外上市销售的生物制品。 2.单克隆抗体。 3.基因治疗、体细胞治疗及其制品。 4.变态反应原制品。
抗体偶联药物
抗体偶联药物(ADC)的涅槃重生 抗体偶联药物(antibody-drug conjugate, ADC)是将抗体与细胞毒性药物连接起来,通过抗体的靶向作用将细胞毒药物靶向肿瘤,进而降低化疗中常见的药物非特异性的全身毒性。抗体偶联药物(antibody-drug conjugate, ADC)的研究可以追溯到1980s,,但是直到2000年,首个抗体偶联药物gemtuzumab o zogamicin(商品名Mylotarg,Pfizer研发)才被FDA批准用于治疗急性粒细胞白血病,但由于偶联技术、靶向性、有效性等受限,完整的抗体偶联药物在血液不稳定,导致致死性毒性的产生,于2010年撤市。这使得本就不明朗的ADC药物研究,更蒙上了一层阴影。 但是随着Takeda/Seattle Genetics 通过对原有技术的改进,利用自己的新型抗体偶联技术开发了brentuximab vedotin(SGN-35,商品名Adcetris,)新型抗体偶联药物,并与2011年被FDA批准用于治疗霍奇金淋巴瘤和系统性间变性大细胞淋巴瘤。2013年抗体偶联药物再次取得突破,Genentech/ImmunoGen 联合开发的Ado-trastuzumab emtansine(T-DM1,商品名Kadcyla)被FDA批准用于HER2阳性乳腺癌,这是首个针对实体瘤的抗体偶联药物。随着这两个药物的研发成功,ADC药物再次以火热的状态进入人们的研究视野。 1、进入临床阶段ADC药物 截至目前大概有30多种ADC药物进入临床开发阶段(表1),统计表中30种药物针对适应症发现,其中仅有4种药物针对实体瘤。主要原因:抗体难于透过毛细管内皮层和穿过肿瘤细胞外间隙到达实体瘤的深部。而使用抗体片段,如Fab,制备分子量较小的偶联物,可能提高对细胞外间隙的穿透性,增加到达深部肿瘤细胞的药物量。因此“抗体的小型化或适度的小型化将会是研制ADC药物的重要途径”。同时我们还能看到ImmunoGen、Seattle Genetics在现有ADC 药物研发中占有绝对的统治地位,这得力于他们成熟的抗体偶联技术——利用天然抗体自身的赖氨酸和半胱氨酸中的巯基偶联药物(non-specific)。 2、如何才能成功开发出一种ADC药物?
189上市前药物临床安全性评价与风险评估(三)--建立上市前安全性数据库的考虑因素
发布日期 20071129 栏目化药药物评价>>化药质量控制 标题上市前药物临床安全性评价与风险评估(三)--建立上市前安全性数据库的考虑因素 作者杨焕 部门 正文内容 审评五部杨焕 摘要:风险评估即患者获益与风险比最大化的管理思路应贯穿于药物的整个生命周期。任何一个新药的风险评估都涉及到数量和质量两个方面,数量是指安全性数据库的规模;质量是指临床试验设计、实施、结果分析全过程的质量,在安全性方面具体是指对于药物不良反应报告、归类、判断、分析总结的质量。本系列文章从技术评价角度,在分析了解国内外药物不良反应监测的历史与发展的基础上,对上市前安全性数据库的规模大小和如何保证安全性数据库的质量方面问题进行了探讨,同时也参考和介绍了国外发达国家最新的风险评估和风险控制理念,其目的是为临床试验的研究者和注册申请人提供参考和建议。 虽然合理的安全性数据库的特征是针对每个新药特有的,但也有一些通用的原则。一般情况下,保证安全性数据库的质量和完整性的工作应当与支持有效性同样的重要。因为对安全性进行评价时,往往要考查来自多个试验中的数据,所以特别要注意检查所用的不良反应相关的术语、评价方法以及标准术语的使用(建议使用医学用语词典-药物管理事务词典MedDRA),以确保安全性信息清晰,准确。对研究期间停用所指定治疗的原因(如任何原因的死亡或脱落)进行确认和分析对于全面了解一个新药的安全性特性非常重要。 申请人和研究者在为新药的临床计划提出具体的临床试验方案时应当考虑下列要素,以利于对临床计划进行充分的风险评估。
(一) 长期对照的安全性研究 在许多新药开发的整体临床计划中,常见多数受试者暴露的临床数据和几乎所有的长期暴露数据都是来自单组(single-arm study)或非对照的研究。虽然这些数据有参考价值,但有些情况下最好是获得有对照的长期安全性数据。这样的数据可以对不良事件/不良反应的发生率进行比较,利于准确判断不良事件/不良反应的发生原因。对照组具体使用阳性/活性对照药或者用安慰剂取决于所治疗的疾病,即对照药选择安慰剂还是阳性对照药,取决于所治疗的疾病在伦理学和医学上的可行性。 1. 阳性对照药的价值取决于所关注的不良事件/不良反应 一般情况下,发生罕见的严重不良事件/不良反应(如重度肝损伤或再生障碍性贫血等)一旦这些事件1)有明确记录,并且2)没有其他原因可以解释,都被认为是重要和有临床意义的,因为这些罕见的严重不良事件/不良反应在任何规模的人群中预期发生率基本都为零,因此,一般这种严重不良事件都能得到合理的解释,无论是否有对照组,都应被看作是值得关注的风险信号。 新药临床试验中,只要发现出现1例某种严重的ADR ,都应在后期陆续的临床研究中对此类ADR加以关注。甚至会反映在将来的药物使用说明书中,期望引起警惕。因此,ADR 的发现率(概率)尤为重要,特别是对于早期研究的Ⅱ期临床试验。 另一方面,在治疗的人群中要检出较常见的不良事件/不良反应发生率的升高情况,就需要设对照组(如缺血性心脏病患者中的猝死)。如果不良事件/不良反应被认为是所治疗的疾病的表现或症状(如哮喘吸入治疗中发生的哮喘急性加重)时,设置对照组就显得特别重要。 因此,对于决定一个新药何时应当进行长期对比性安全性研究,应当根据该产品将来的适应症,即说明书中所治疗的目标患者人群(如果严重不良事件的发生率比较高,就更为有用),以及早期所进行的临床和临床前安全性评价。尽管不会经常进行长期有对照的临床安全性研究,但如果早期开发中发现了药物的安全性问题,则此类研究就会特别有用。这些情况下,可能比较适于采用为检验特定风险问题的假设而设计的安全性研究,随着累积暴露量的增加,就会有更多机会发现和检出所关注的安全性问题。
在研抗体偶联药物及市场分析
第3弹:在研抗体偶联药物及市场分析 1.上市抗体偶联药物 抗体偶联药物(antibody-drug conjugate, ADC)的研究可以追溯到1980s,将抗体与细胞毒药物偶联产生协同作用,同时通过抗体将药物直接输送到靶细胞。然而早期产品的临床效果并不尽如人意,由于偶联技术、靶向性、有效性等受限,完整的抗体偶联药物在血液不稳定。偶联技术的发展以及诸多靶标的发现催生了第二代抗体偶联药物,它们在血液中的稳定性有了很大提高,足以将细胞毒药物输送到靶细胞。 抗体偶联药物取得突破性进展是在2011年,FDA批准了CD30 特异性的Adcetris (brentuximab vedotin, SGN-35)用于治疗霍奇金淋巴瘤(Hodgkin’s lymphoma)和系统性间变性大细胞淋巴瘤(systemic anaplastic large cell lymphoma)。该药由三部分构成:嵌合IgG1 抗体cAC10+微管聚合抑制剂MMAE (Monomethyl auristatin E)+可被蛋白酶裂解的连接子,cAC10能够特异性识别CD30,MMAE则起到杀死肿瘤细胞的作用。该药由Millennium (The Takeda Oncology Group)和Seattle Genetics共同研发,享有专利保护的连接子和偶联技术(cytotoxic platform technology)出自Seattle Genetics公司。由于病人数量相对较少,该药在美国的年销售额(2011-10至2012-09)为1.36亿美元。 2013年2月,Genentech研发的Kadcyla (ado-trastuzumab emtansine, T-DM1)获得FDA批准,用于治疗HER-2阳性转移性乳腺癌,III临床研究显示Kadcyla优于拉帕替尼+卡培他滨。Kadcyla也是由三部分构成:曲妥珠单抗+微管聚集抑制剂DM1+连接子,曲妥珠单抗靶向HER2,本身也已被批准治疗乳腺癌,DM1是天然产物Maytansine衍生物,能够与微管花位点结合,产生细胞毒作用。DM1的细胞毒作用比标准化疗高100-10000倍,由于曲妥珠单抗对肿瘤细胞具有选择性,该药的毒性比单纯使用DM1低。该药的连接子及偶联技术(Targeted Antibody Payload ADC technology)出自ImmunoGen公司,Genentech 获得了其专利许可。 2.在研抗体偶联药物 抗体偶联药物的核心是偶联子及偶联技术(ADC platform technology),而主导这项技术的两家公司是Seattle Genetics和ImmunoGen,各大公司的在研项目多与在这两家公司的合作下展开,Genentech和Pfizer自己也在投资做前期研究。在研抗体偶联药物: 3.市场分析 两种利益驱动着抗体偶联药物的研发,一是通过这种方式产生新的专利,二是抗体药
药物安全性评价实验
安全性评价实验 实验材料及动物 受试物:五味消毒饮分散片 实验动物:清洁级昆明小鼠100只 急性毒性试验方法 取小鼠100 只,随机分为10 组,每组10 只,雌雄各半,禁食12 h 后分别称重。根据改进寇氏法设9个剂量组,由低到高的组间剂量比为0175 :1 ,第Ⅰ~Ⅸ组的给药剂量分别以28.88 ,38.51 , 51.34 , 68.46 , 91.28 , 121.70 , 162.27 , 216.36, 和288.48 g·kg- 1 ,以0.75 稀释等级系数,第Ⅰ~Ⅷ组用蒸馏水稀释成相等的容量2.4 mL 灌胃, Ⅸ组为五味解毒水煎剂原液2.4 mL 灌胃,第Ⅰ~Ⅸ组的药物配制浓度分别 0.24 ,0.32 ,0.43 ,0.57 ,0.76 ,1.01,1.35 ,1.80 ,2.40 g·mL- 10. 每次灌胃体积均为20 mL·kg- 1 ,每次间隔4 h ;第Ⅹ为对照组,灌服等量蒸馏水。每天观察记录动物的毒性反应及死亡情况,对死亡动物及时剖检、记录病理变化,连续观察7 d。根据各组小鼠死亡数,再计算LD50。 亚慢性毒性试验方法 动物分组处理取小鼠80只,随机分为4 组,每组20只,雌雄各半。Ⅰ、Ⅱ、Ⅲ组为药物的高、中、低剂量组,分别以LD50的四分之一,十分一,二十五分之一剂量灌胃,每次灌胃体积均为20 mL·kg - 1 ; Ⅳ组为对照组,每日定时灌胃等量蒸馏水。各组连续灌胃14 d ,停药后
再持续观察14 d。 观察指标临床表现每天观察小鼠的一般状况,每周测定体重变化情况,死亡小鼠及时剖检。 血常规变化于停药后第1天和第14 天,各组随机抽取10 只采血, EDTA 二钾盐抗凝,立即用AC·Tdiff2 型全自动血细胞计数分析仪测定红细胞数、白细胞总数、血小板数和血红蛋白含量。 血清生化值变化于停药后第1 天和14 天,每组随机抽取10 只采血,分离血清,用Vitalab Selecctra E自动生化分析仪测定谷丙转氨酶、谷草转氨酶、碱性磷酸酶、尿素氮、肌酐、总蛋白、白蛋白的含量。 病理学变化于停药后第1 天每组随机抽取3只,停药后第14 天对剩余所有小鼠剖检,采集心、肝、脾、肺、肾、胃和肠,固定,石蜡切片,HE 染色,显微镜下观察病理变化。 通过急性毒性实验,得出五味消毒饮分散片在相应的剂量中,未引起小鼠的死亡,可以认为五味消毒饮分散片在剂量范围内未引起毒性反应。 亚慢性毒性实验期间,观察各组小鼠生长发育是否良好,各种体征表现是否正常。五味消毒饮分散片提取物对动物体重、脏器重量、血液谷丙转氨酶(ALT)、谷草转氨酶(AST)、尿素氮(BUN)、肌醉(Cr)、血清白蛋白(Alb)和血糖(Glu)均有无显著影响。 长期毒性试验 受试物
干货抗体偶联药物(adc)深度研究报告
干货抗体偶联药物(adc)深度研究报告 目录一、行业背景 针对病症 历史沿革基本原理介绍二、核心技术抗体部分(1)靶点选择(2)抗体选择(3)抗体修饰(4)抗体内吞连接物部分(1)连接物(Linker)选择(2)连接方式及DAR(3)新技术Abzena的ThioBridge毒素部分核心专利-连接物与毒素排序三、效果对比:Kadcyla对比Herceptin四、核心公司竞争情况 1. 领先公司(1)Seattle Genetics (2)ImmunoGen (3)Immunomedics 2. 规模较大公司 (1)Abzena (2)Agensys (3)Celldex (4)Progenics Pharmaceuticals (5)Genmab (6)Sorrento旗下Concortis 3. 大型药企在ADC领域的布局(1)Abbvie及其旗下Stemcentrx (2)Roche旗下Genetech的情况(3)武田旗下Takeda Oncology (4)辉瑞ADC产品Mylotarg (5)复星、药明、浙江医药与Ambrx (6)三生制药及三生国健(7)丽珠医药集团旗下丽珠单抗(8)江苏恒瑞医药(9)四川恒康旗下上海美雅珂生物(10)其他药企五、行业市场规模六、ADC成功要素分析独家技术优秀团队研发方向
一、行业背景抗体偶联药物Antibody-drug Conjugate (ADC) 是拥有强细胞毒性的化疗药物通过连接物与单抗偶联形成的,兼具小分子药物强大的杀伤力和纯单抗高度的靶向性,因而成为肿瘤靶向治疗的研究和发展热点。但是,ADC 本身并非在各方面强于纯单抗。其疗效的显著提升是通过牺牲药品的均一性与稳定性实现的。现在比较成熟的两种偶联技术分别侧重均一性与稳定性,有一些新式偶联技术能够在两方面同时改善。1、针对病症ADC药物被用于癌症治疗,其针对病症由其中抗体所针对的靶点决定,能够对将该靶点高表达的肿瘤细胞进行针对性DNA破坏或抑制微管。由于其针对性很高,其可以使用化疗中不能使用或剂量不能提高的高毒性药物[1]。ADC药物相对于化疗药的治疗安全窗口therapeutic window会更大,相对更加安全[2]。下表为可供使用的各种靶点及其针对的癌症种类。其中,目前已经上市的两款药物中,Kadcyla使用HER2靶点,针对HER2阳性的肺癌;Adcetris使用CD30靶点,针对CD30阳性的霍奇金淋巴瘤Hodgkin Lymphoma (HL)与间变性大细胞淋巴瘤anaplastic large cell lymphoma (ALCL) [3]。靶向药物中不同抗原及其针对癌症种类,以及相应在研的ADC药物数量抗原对应在研ADC数量主要针对适应症在肿瘤细胞表面表达的靶向抗原GPNMB1乳腺癌及黑素瘤CD561小细胞肺癌(SCLC)TACSTD2
ICH 生物技术药物的临床前安全性评价 S6 R1
人用药品注册技术要求国际协调会 ICH三方协调指导原则 生物技术药物的临床前安全性评价 S6(R1) 1997年7月16日总指导原则 现行第四阶段版本 2011年6月底整合2011年6月12日的附录 本指导原则由相应的ICH专家小组制定,按照ICH进程,已递交管理部门讨论。在ICH进程第四阶段,最终草案被推荐给欧盟、日本和美国的管理机构采纳。
生物技术药物的临床前安全性评价 ICH三方协调指导原则 目录 第I部分: (1) 1.前言 (1) 1.1背景 (1) 1.2目的 (1) 1.3范围 (1) 2.受试物的质量标准 (2) 3.临床前安全性试验 (2) 3.1 一般原则 (2) 3.2生物活性/药效学 (3) 3.3动物种属/模型选择 (3) 3.4动物的数量/性别 (4) 3.5给药途径/剂量选择 (4) 3.6免疫原性 (4) 4.特殊考虑 (5) 4.1安全药理学 (5) 4.2暴露评价 (5) 4.2.1药代动力学和毒代动力学 (5) 4.2.2测定 (6) 4.2.3代谢 (6) 4.3单次给药毒性研究 (6) 4.4重复给药毒性研究 (6) 4.5免疫毒性研究 (7) 4.6生殖能力和发育毒性研究 (7) 4.7遗传毒性研究 (7) 4.8致癌性研究 (7) 4.9局部耐受性研究 (8) 注释 (8) 第II部分: (9) 1.前言 (9)
1.1附录目的 (9) 1.2背景 (9) 1.3指导原则的范围 (9) 2.种属的选择 (10) 2.1一般原则 (10) 2.2一或两个种属 (10) 2.3同源蛋白的使用 (11) 3.研究设计 (11) 3.1剂量选择和PK/PD原则的应用 (11) 3.2研究期限 (11) 3.3恢复 (11) 3.4探索性临床研究 (12) 4.免疫原性 (12) 5.生殖和发育毒性 (12) 5.1一般评论 (12) 5.2生育能力 (13) 5.3胚胎–胎儿发育(EFD)和出生前/后的发育(PPND) (13) 5.4研究的时间安排 (14) 6. 致癌性 (14) 注释 (15) 参考文献 (18)
药物安全性评价word
药物安全性评价word 一、药物安全性评价概述药品:指用于预防、治疗、诊断疾病,有目的地调节人的生理机能并规定有适应症或者功能主治、用法和用量的物质,包括中药材、中药饮片、中成药、化学原料药及其制剂、抗生素、生化药品、放射性药品、血清、疫苗、血液制品和诊断药品等。(《中华人民共和国药品管理法》)(一)新药研发程序 1、探索和筛选:应用常规的药理学方法检测化合物的药理学活性,超过活性标准即可认为有活性;通过早期毒性筛选尽量排除可能有问题的化合物,从而增加测试化合物的数量,减少实验动物的用量和扩大给药剂量的范围。 2、非临床研究:实施符合GLP标准的毒理学安全性评价、生物利用度评价及药代毒代动力学研究。 3、临床研究:Ⅰ期:证明人类对该药物的耐受性并确定其在人体体内的药物代谢动力学特性。Ⅱ期:确定药物的量效关系。Ⅲ期:临床药效学试验(全面、多中心、大量患者参与)Ⅰ期、Ⅱ期、Ⅲ期临床试验应符合GCP标准。 4、申请新药注册:《药品管理法》、《药品注册管理办法》 5、新药上市:进行药物不良反应监测,并进行符合GCP标准的Ⅳ期临床试验 (二)安评的起源最早提出药物安全性评价是缘于20世纪全世界出现了许多严重的药物中毒事件。 1、磺胺酏剂事件(30年代,美国)a、1937年,美国一家公司的主任药师瓦特金斯(HaroldWotkins)为使小儿服用方便,用二甘醇代替酒精做溶媒,配制口服液体制剂,称为磺胺酏剂。b、未做动物实验,在美国田纳西州的马森吉尔药厂投产后,全部进入市场,用于治疗感染性疾病。c、到这一年的9~10月间,美国南方一些地方开始发现患肾功能衰竭的病人大量增加,共发现358名病人,死亡107人(其中大多数为儿童),成为上世纪影响最大的药害事件之一。 2、有机锡中毒事件(50年代,法国)a、烷基锡化合物可引起多起中毒。 b、1954年在法国用于治疗皮肤化脓很有效的Stallion药物,这种药物中含有
新药上市前临床试验安全性数据的分析与评价
464中国临床药理学杂志 第25卷第5期2009年9月(总第12l期) 新药上市前临床试验安全性数据的分析与评价 Clinicalsafetydataanalysisandevaluationofnewdurgsinthepre-approvalphase 杨焕 (国家食品药品监督管理局药品审评中心,北京100038) YANGHuan (CenterforDrugEvaluation,StateFoodandDrugAdministration.Beijing100038,China) 收稿日期:2009—02—17 修回日期:2009—07—14 作者简介:杨焕(1970一),女,副主任医师,主要从事药品技术审评工作 通讯作者:杨焕 Tel:(olo)68585566 E—mail:yanshuman@cde.orgycn摘要:本文分析了上市前药物临床试验进行不良反应监测与评价的特点,介绍了安全性数据的分析、总结和评价方法,阐明了临床试验中需要特别关注和要评价的安全性问题,强调风险评估(即患者获益与风险比最大化)的管理思路应贯穿于药物的整个生命周期中。 关键词:新药;上市前临床试验;安全性数据 中图分类号:R954文献标识码:C 文章编号:1001—6821(2009)05—0464—03 关于临床试验中的安全性数据如何进行总结、分析与评价,自20世纪90年代以来,人用药品注册技术要求国际协调会(ICH)就相继发布了药品注册的国际技术要求,如ICH中的E2/E3/E6/E9,均涉及此方面的内容¨o;国家食品药品监督管理局(SFDA)在2005年也相继发布了《化学药物临床试验报告的结构与内容技术指导原则》和《化学药物和生物制品临床试验的生物统计学技术指导原则》心-3],建议同时参考。 1上市前临床药物不良反应监测与评价的特点 上市前的临床试验是对药物的有效性、安全性进行科学的评价,是新药开发的重要环节,为国家药品监督管理部门批准其生产上市提供依据。《药品I晦床试验管理规范》(GCP)是指导和规范药物临床试验过程的法规性文件MJ,可以有效地保证临床试验结果的科学可靠,保护受试者的权益和安全。上市前药物临床试验中的安全性评价作为关键环节,同样必须遵循GCP的要求。 1.1GCP中的有关安全性评价的要求 GCP中的临床试验方案、研究者职责、申办者(注册申请人)职责、监查员的职责、记录与报告等章节,都包含对安全性数据收集和管理(即药物不良反应监测)的具体要求,由此可见对药物安全性评价的要求,贯穿整个GCP的实施中。 1.2药物上市前不良反应监测的特点 药物不良反应监测的特点有以下5种:①在新药临床试验期问,因用药单一并用于特定目标人群和针对惟一的适应证,对于出现的不良事件较好归因。②药物不良反应定义有所不同,上市前临床研究中,由于正在进行研究的试验药物的用药剂量、疗效等均未完全确定,因此,任何剂量下出现的与药物有关的、有害的且非期望的反应,都应当视为药物不良反应,这与世界卫生组织(WHO)对于上市后药物不良反应的定义有一定的差异。③上市前临床试验中的不良反应/不良事件(ADR/AE)报告,均来自有目的、明确的前瞻性临床研究,这使得ADR/AE关联性评价较上市后易于判断。④上市前临床试验中,更强调对个例严重且非预期不良反应的快速报告和评价。 万方数据
在研抗体偶联药物及市场分析
第3弹:在研抗体偶联药物及市场分析 1?上市抗体偶联药物 抗体偶联药物(antibody-drug conjugate, ADC )的研究可以追溯到1980s,将抗体与细胞毒药物偶联产生协同作用,同时通过抗体将药物直接输送到靶细胞。然而早期产品的临床效果并 不尽如人意,由于偶联技术、靶向性、有效性等受限,完整的抗体偶联药物在血液不稳定。 偶联技术的发展以及诸多靶标的发现催生了第二代抗体偶联药物,它们在血液中的稳定性有 了很大提高,足以将细胞毒药物输送到靶细胞。 抗体偶联药物取得突破性进展是在2011年,FDA批准了CD30特异性的Adcetris (bre ntuximab vedoti n, SGN-35)用于治疗霍奇金淋巴瘤( Hodgkin 'lymphoma )和系统性间变性大细胞淋巴瘤 (systemic anaplastic large cell lymphoma )。该药由三部分构成:嵌合IgG1抗体CAC10+微管聚合抑制剂MMAE (Monomethyl auristatin E)+可被蛋白酶裂解的连接子,cAC10 能够特异性识别CD30, MMAE 则起到杀死肿瘤细胞的作用。该药由Millennium (The Takeda Oncology Group)和Seattle Genetics共同研发,享有专利保护的连接子和偶联技术( cytotoxic platform technology)出自Seattle Genetics公司。由于病人数量相对较少,该药在美国的年销售额(2011-10至2012-09 )为1.36亿美元。 2013 年2 月,Genentech 研发的Kadcyla (ado-trastuzumab emtansine, T-DM1)获得FDA 批准,用于治疗HER-2阳性转移性乳腺癌,III临床研究显示Kadcyla优于拉帕替尼+卡培他滨。Kadcyla也是由三部分构成:曲妥珠单抗+微管聚集抑制剂DM1 +连接子,曲妥珠单抗靶向 HER2,本身也已被批准治疗乳腺癌,DM1是天然产物Maytansine衍生物,能够与微管长春 花位点结合,产生细胞毒作用。DM1的细胞毒作用比标准化疗高100-10000倍,由于曲妥 珠单抗对肿瘤细胞具有选择性,该药的毒性比单纯使用DM1低。该药的连接子及偶联技术(Targeted Antibody Payload ADC technology )出自ImmunoGen 公司,Genentech 获得了其专利许可。2?在研抗体偶联药物抗体偶联药物的核心是偶联子及偶联技术( ADC platform tech no logy ),而主导这项技术的 两家公司是Seattle Genetics和ImmunoGen,各大公司的在研项目多与在这两家公司的合作下展开,G e n e n t e c h和P f i z e r自己也在投资做前期研究。在研抗体偶联药物: (partner^Product mD匚Lead Nk>tabUvi?b R enntiy k unched R CM hTGtnittiiiH li III mriuri 托 日1H? | T-CHM 11H ER Pf; rt-tdfic: mAli LhiLml LoDMl EMILIA. MARIANNE. 加 K J THERESA Ahdse Nil (CMC-1! 441to caiich?arriictfi Nofl「Hodiqfc iiti\ |卉吋乩 ALL — Phu無 1 nn^UuFitjmiah mcHliiirrH nc FlMGN QOll sprrrtir mA h linked to DM 1Sriwllrril rnulTir^r^c nFma |卩。NORTH CiHldf at霑空Glf^ihd! Lirtrk|j|1Mh udAtJriitCDX-Oll)CRN MB ?ilU mAh hnif-d 诂 MMAE Haq 占1 I^IV EMERGE R CM111?■'Giri iwilw h iSBdLlkf 幼RGrJSOT urid tcurrabirulKjrb wiLhrltUklllidb) 匚DM叫严皿匸(RG-7悶 > 柯 CD 皿*die帆G-巧弼]mAb lir-ktHj toMMAE or MMAf FdlliLuiur lyuiphiiiiT]軌 DLBCLCLLiP^ Prc?yui Fiidrsr^-d |S?Llie 匸f tiL# PSMA.ADC P SMA^peciriL niAii linked toMMM Q-tdllLeH一 barxrfi llmrTiurioGcn*W4W CD LS-sprcWit rnAb Nn ked to[)U4 DLBCLBffillALL二 3?市场分析 两种利益驱动着抗体偶联药物的研发,一是通过这种方式产生新的专利,二是抗体药物协同作用产生的高额溢价。使用Adcetris每年的治疗费用在100000美元,而使用Kadcyla 每月的治疗费用也高达9800美元。