克隆载体表达载体构建详细版
表达载体的构建方法及步骤

For personal use only in study and research; not for commercial use表达载体的构建方法及步骤一、载体的选择及如何阅读质粒图谱目前,载体主要有病毒和非病毒两大类,其中质粒DNA 是一种新的非病毒转基因载体。
一个合格质粒的组成要素:(1)复制起始位点Ori 即控制复制起始的位点。
原核生物DNA 分子中只有一个复制起始点。
而真核生物DNA 分子有多个复制起始位点。
(2)抗生素抗性基因可以便于加以检测,如Amp+ ,Kan+(3)多克隆位点MCS 克隆携带外源基因片段(4)P/E 启动子/增强子(5)Terms 终止信号(6)加poly(A)信号可以起到稳定mRNA 作用选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。
如果构建的目的是要表达一个特定的基因,则要选择合适的表达载体。
载体选择主要考虑下述3点:【1】构建DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。
【2】.载体的类型:(1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小)。
如<10kb 选质粒。
(2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。
(3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。
【3】载体MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产生阅读框架错位。
综上所述,选用质粒(最常用)做载体的5点要求:(1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载体);(2)一般使用松弛型质粒在细菌里扩增不受约束,一般10个以上的拷贝,而严谨型质粒<10个。
(3)必需具备一个以上的酶切位点,有选择的余地;(4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位Ampr(试一试)。
克隆载体与表达载体

克隆载体与表达载体
1. T载体是克隆载体,你的基因通过TA克隆法插入载体,这一步的目的是扩增基因,得到大量你要的目的基因片段,以便进行下一步表达载体的构建;DH5α是克隆菌株,不能用来做表达;
2. 欲在大肠杆菌中表达外源基因,需要首先构建原核表达载体,如可将构建至T 载体的目的通过双酶切切下来然后连接到表达载体上,如PET系列的载体等,构建成原核表达载体后,可将此载体转化表达菌株,如BL21(DE3,)等,如果你的目的基因含有稀有密码子,也可以转化Rosetta系列的表达菌株。
最后将构建成功的基因工程菌进行诱导表达。
1、T载体常用于克隆,一般来讲都会再把目的基因亚克隆到表达载体上。
但是并非T载体不能用来表达。
常见的pMD18-T,含有lacZ操纵子,可以IPTG诱导表达。
pGEM-T则含有T7和SP6启动子。
2、为了能够顺利地使用T7系统来表达蛋白,在如BL21(DE3)一类的大肠杆菌菌株中,编码T7RNA聚合酶的基因被整合到其染色体上,并位于lacUV5启动子的下游,受乳糖操纵子调控。
而目标蛋白的编码序列则被构建到含T7启动子序列的质粒上,并受T7RNA聚合酶调控转录。
3、构建质粒、基因表达看似比较成熟,也比较简单。
其实这里面大有学问。
学会看菌株和质粒的相关文档,答案就在其中。
表达载体的构建方法及步骤

令狐采学创作表达载体的构建方法及步骤令狐采学一、载体的选择及如何阅读质粒图谱目前,载体主要有病毒和非病毒两大类,其中质粒DNA是一种新的非病毒转基因载体。
一个合格质粒的组成要素:(1)复制起始位点(加即控制复制起始的位点。
原核生物DNA分子中只有一个复制起始点。
而真核生物DNA分子有多个复制起始位点。
(2)抗生素抗性基因可以便于加以检测,如Amp+ ,I<an+(3)多克隆位点MCS克隆携带外源基因片段(4)P/E启动子/增强子(5)Terms终止信号(6)加poly (A)信号可以起到稳定mRNA作用选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。
如果构建的目的是要表达一个特定的基因,则要选择合适的表达载体。
载体选择主要考虑下述3点:[1]构建DNA重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。
【2】.载体的类型:令狐采学创作令狐采学创作(1)克隆载体的克隆能力一据克隆片段大小(大选大,小选小)。
如<n)kb选质粒。
(2)表达载体据受体细胞类型一原核/真核/穿梭,E.coli/哺乳类细胞表达载体。
(3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位; 表达天然蛋白质或融合蛋白作为相应载体的参考。
【3】载体MCS中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产主阅读框架错位。
综上所述,选用质粒(最常用)做载体的5点要求:(1)选分子量小的质粒,即小载体(l-1.5kb) 一不易损坏,在细菌里面拷贝数也多(也有大载体);(2)一般使用松弛型质粒在细菌里扩增不受约束,一般1()个以上的拷贝,而严谨型质粒<1()个。
(3)必需具备一个以上的酶切位点,有选择的余地;(4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位Ampr (试一试)。
(5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Pur。
克隆载体表达载体构建详细版

克隆载体表达载体构建详细版一、稀释引物1、4℃,15min、13000转离心(先等离心机降温)2、根据OD值加DD水。
3、静置30min(冰上)4、准备1.5毫升EP管,并加90ulDD水。
5、向EP管中加10ul引物,震荡离心,-20℃保存。
二、跑MIX检测引物(20ul体系)、上引物0.8ul下引物0.8ulMix 10ulDNA(日本晴)1ulDD水7.4ul三跑高保真酶(50ul体系)DNAorCDNA 2ul上引物2ul下引物2ul5*buffer 10uldNTPs 5ulDD水28ulPfu(最后加)1ul四胶回收流程1、在紫外线下切胶,用牙签装入2ml的EP管中。
2、按量加XP2,放在55℃水浴锅中10min,每2min摇匀1次,涡旋,短离。
3、将液体冷却到室温,转移到平衡住中,离心10000转,1min30s,倒掉滤液。
4、加入xp2 300ul,离心10000r,1min30s,倒掉滤液。
5、加入spw700ul,离心10000r,1min,倒掉滤液(重复一次)6、空转2min,13000r,之后换1.5mlEP管。
7、套上保鲜膜放入37℃烘箱中,30min。
8、加入DD水10ul,静置2min,离心2min,13000r,重复3次,-20℃保存。
五、胶回收产物检测(10ul)体系上引物0.4ul下引物0.4ulMix 5ul回收产物1ulDD水 3.2ul六、构建blunt cloning 载体(克隆载体)(4ul 体系)胶回收产物 3.5ulBlunt cloning 0.5ul混匀后,PCR:25℃15min 盖子温度50℃之后转化1、提前5min从-70℃冰箱中拿出大肠杆菌感受态,冰上解冻5min。
2、将样品(4ul)加入感受态的大肠杆菌中,冰上30min,大约剩5min左右,打开水浴锅预热到42℃,并拿出SDC 培养基解冻(室温解冻)。
3、水浴锅42℃,60-90s迅速转移到冰浴2min,该过程中不要摇动离心管。
构建表达载体的一般流程

构建表达载体的一般流程
构建表达载体的一般流程如下:
1. 选择合适的载体:根据需要表达的基因或蛋白质,选择一个合适的表达载体。
常用的载体有质粒、病毒、合成RNA等。
2. 插入目标基因:将需要表达的基因插入到载体的多克隆位点中。
可以通过PCR 扩增目标基因并使用限制性内切酶切割载体和基因,然后进行连接。
3. 连接启动子和终止子:为确保基因在宿主细胞中能够正常表达,需要在基因的上游添加启动子和在基因的下游添加终止子。
这些序列将调控基因的表达方式和水平。
4. 检验构建质量:通过限制性内切酶切割、PCR扩增或测序等方法,验证所构建的载体和目标基因的正确性和完整性。
5. 转化宿主细胞:将构建好的表达载体导入到宿主细胞中。
常用的转化方法有电穿孔、热激冲击和化学转化等。
6. 筛选和鉴定:通过特定的筛选方法或选择标记,在转化的宿主细胞中筛选和鉴定表达目标基因的阳性克隆。
7. 表达优化:根据需要,可以通过调节培养条件、添加诱导剂或使用特定的宿主菌株等方法,优化目标基因的表达水平和质量。
8. 收获表达产物:经过一段时间的培养后,收获表达的基因产物,如蛋白质,进行后续的纯化、鉴定和应用等。
需要注意的是,具体的构建流程和方法可能会因实验的目的和要求而有所不同。
在实施过程中,需要对不同步骤和条件进行优化和调整,以确保表达载体的构建和表达目标基因的成功。
分子克隆详细步骤

分子克隆详细步骤分子克隆是通过重组DNA分子来产生大量完全相同的DNA序列的技术。
在分子克隆工作中,我们主要进行克隆载体的构建、目标DNA的扩增、将目标DNA插入克隆载体中、转化和筛选等步骤。
下面将详细介绍这些步骤:1.克隆载体的构建:克隆载体是用于插入目标DNA的DNA分子。
常用的克隆载体包括质粒、噬菌体和人工染色体等。
在构建克隆载体时,我们首先需要选择适合的载体,并提取载体的DNA。
然后,利用酶切酶对载体进行酶切,生成线性的载体DNA。
接下来,将目标DNA插入克隆载体的相应位点上,形成重组的载体。
2.目标DNA的扩增:目标DNA可以通过PCR(聚合酶链反应)来扩增。
首先,设计引物,使其与目标DNA的两端末端相互互补。
然后,在PCR反应中,通过DNA聚合酶的扩增作用,使目标DNA得以扩增。
PCR反应通常包括模板DNA、引物、核苷酸和聚合酶等成分。
3.目标DNA的插入:将扩增后的目标DNA与酶切后的载体进行连接,利用DNA连接酶催化目标DNA与载体之间的连接反应,生成重组的克隆载体。
连接后的载体含有目标DNA的序列。
4.转化:将克隆载体引入宿主细胞中进行复制。
这一步骤通常称为转化。
转化可以通过电击、热激、化学方法等方式进行。
宿主细胞通常是大肠杆菌等细菌。
5.筛选:利用筛选方法来选择包含目标DNA的克隆。
常用的筛选方法包括抗生素筛选、报告基因筛选和限制性内切酶酶切筛选等。
抗生素筛选是将带有选择性抗生素耐受基因的克隆引入含有相应抗生素的培养基中,只有带有目标DNA的克隆才能生长。
报告基因筛选是通过将报告基因插入克隆载体中,使之与目标DNA一起被转录和翻译,从而表达报告基因的蛋白质,以此来筛选包含目标DNA的克隆。
限制性内切酶酶切筛选是通过限制性内切酶对重组载体和目标DNA进行酶切,并通过凝胶电泳的方法来分离并检测含有目标DNA的克隆。
以上就是分子克隆的详细步骤。
通过这些步骤,我们可以获得大量完全相同的DNA序列,并用于各类分子生物学研究和应用中。
克隆载体与表达载体
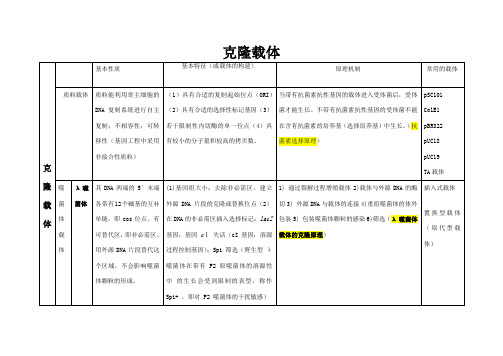
克隆载体
基因间隔区(intergenic region, IG 区)基因II与基因IV之间存在一段507bp的基因间隔区,内含有复制起始位点,是实施改造、构建人工载体的重点区域。
② IG区内只有一个Bsu I 切点。
(2)加入酶切位点,在IG区内加入单一内切酶位点。
M13mp1 在IG区内插入一个大肠杆菌的LacZ’(-肽序列)。
使克隆的DNA片段以特定单链的形式输出受体细胞外,M13重组分子筛选简便,被M13噬菌体感染的受体细胞生长缓慢,形成混浊斑,易于辨认挑选。
而且重组分子越大,混浊斑的混浊度亦越大但M13-DNA载体的最大缺陷是装载量小,只有 kb
考斯质粒是一类人工构建的含有λ-DNA cos序列和质粒复制子的的特殊类型载体。
能像
-DNA那样进行体外包装,并高效转染受体细胞;能像质粒那样在受体细胞中自主复制具有较高容量的克隆能力:45kb;具有与同源性序列的质粒进行重组的能力粘粒(cosmid)是带有 cos 序列的质粒。
cos序列是噬菌体 DNA 中将DNA 包装到噬菌体颗粒中所需的 DNA 序列。
黏粒的组成包括质粒复制起点(colE1),抗性标记(amp r),cos 位点,因而能象质粒一样转化和增殖。
克隆的最大 DNA 片段可达 45kb 。
有的粘粒载体含有两个cos 位点,在某种程度上可提高使用效率。
质粒载体总结
λ噬菌体载体
表达载体。
克隆载体与表达载体介绍

(二)质粒载体的构建
天然质粒载体不易直接作为克隆载体,需要改造:
(1)选择合适的复制起始位点,松散型质粒复制起始位点; (2)加入合适的选择标记基因,主要有LacZ基因和抗生素抗性基因; (3)增加或减少酶切位点,组装多个单一的限制酶切位点即多克隆位点
(MCS); (4)缩短长度,通常重组DNA分子越小,转化效率越高。
(三)常用的质粒载体
1. pBR322质粒载体
2. pUC18和pUC19质粒载体
3. TA载体
二、噬菌体载体
(一) λ 噬菌体载体 1 λ 噬菌体的性质
λ噬菌体Βιβλιοθήκη 外壳蛋白 线性,全长48502bp
λDNA 5’端含12bp的黏性末端
2 λ 噬菌体的构建 构建λ 噬菌体的依据:
a.λ 噬菌体能够包装原λDNA长度的75%-105%的DNA片段 (36.4~51kb) b.有20kb的区域对λ噬菌体生长非必需。
该λ 噬菌体载体的最大装载容量为: 4.9+5.5+2.6+(51-48.5)=15.5kb
2.6kb
3 代表性λ 噬菌体
(1) 插入型载体
装载容量为:0-10.18kb
(2) 置换型噬菌体载体载体 克隆外源DNA片段范围为:9-23kb
(二) M13 噬菌体载体
1.M13噬菌体的增值
M13噬菌体是单链丝状噬菌体; 长6407bp, 507bp基因间隔区,含复制起始位点,同时可用于改造; M13 DNA在宿主细胞中以双链或单链形式存在,但释放到细胞外的 M13 噬菌体颗粒以单链形式存在。
2.M13噬菌体载体 1) 插入选择标记基因; 2) 组装合适的多克隆位点。
三、噬菌体-质粒杂合载体
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
一、稀释引物
1、4℃,15min、13000转离心(先等离心机降温)
2、根据OD值加DD水。
3、静置30min(冰上)
4、准备1.5毫升EP管,并加90ulDD水。
5、向EP管中加10ul引物,震荡离心,-20℃保存。
二、跑MIX检测引物(20ul体系)、
上引物0.8ul
下引物0.8ul
Mix 10ul
DNA(日本晴)1ul
DD水7.4ul
三跑高保真酶(50ul体系)
DNAorCDNA 2ul
上引物2ul
下引物2ul
5*buffer 10ul
dNTPs 5ul
DD水28ul
Pfu(最后加)1ul
四胶回收流程
1、在紫外线下切胶,用牙签装入2ml的EP管中。
2、按量加XP2,放在55℃水浴锅中10min,每2min摇匀1次,涡旋,短离。
3、将液体冷却到室温,转移到平衡住中,离心10000转,1min30s,倒掉滤液。
4、加入xp2 300ul,离心10000r,1min30s,倒掉滤液。
5、加入spw700ul,离心10000r,1min,倒掉滤液(重复一次)
6、空转2min,13000r,之后换1.5mlEP管。
7、套上保鲜膜放入37℃烘箱中,30min。
8、加入DD水10ul,静置2min,离心2min,13000r,重复3次,-20℃保存。
五、胶回收产物检测(10ul)体系
上引物0.4ul
下引物0.4ul
Mix 5ul
回收产物1ul
DD水 3.2ul
六、构建blunt cloning 载体(克隆载体)(4ul 体系)
胶回收产物 3.5ul
Blunt cloning 0.5ul
混匀后,PCR:25℃15min 盖子温度50℃
之后转化
1、提前5min从-70℃冰箱中拿出大肠杆菌感受态,冰上解冻5min。
2、将样品(4ul)加入感受态的大肠杆菌中,冰上30min,大约剩5min左右,打开水浴锅预热到42℃,并拿出SDC 培养基解冻(室温解冻)。
3、水浴锅42℃,60-90s迅速转移到冰浴2min,该过程中不要摇动离心管。
4、加入900ulSOC培养基,混匀。
5、37℃,160r摇床1h,使细菌复苏。
6、取出后离心3000r,2min。
7、吸出上清,留下100-150ul左右冻在LB 100ml+kana或者A 固体培养基上。
8、涂板,37℃过夜培养。
9、挑单菌落(一般不超过7个)加入到10ul的DD水中。
菌液PCR体系(10ul体系)
上游引物0.4ul
下游引物0.4ul
Mix 5ul
菌液1ul
DD水 3.2ul
之后跑电泳,若有目的条带则继续。
七摇菌
菌液+LB液体(加抗生素)-------L管(过夜摇菌)
第二天500ul菌液+500ul甘油混匀保存2管,其中一管送检,另一管-70℃保存。
若检测结果正确
八、提取质粒
1、在1.5ul的EP管中收集菌液,10000转,1min,弃上清,重复一次。
2、加205ul solution I(提前预热),用枪头打匀,室温静置2min。
3、加250ul solution II(提前预热),混匀,室温静置2min。
4、加350ul solutionIII,混匀,室温静置2min(有白色丝状物产生)
5、13000转,10min,取700ul上清液于吸附柱中,10000转,1min。
6、加500ul HBbuffer,12000转,1min,倒掉废液。
7、加700ul DNA wash buffer,13000转,1min,倒掉废液,重复一次。
再空离心2min 13000转。
8、37℃烘箱中干燥5分钟,加50ulDD水溶解,静置2min,离心13000转,2min。
九做酶切(50ul)
DD水19ul
K-B 5ul
质粒20ul
bamI 3ul
HIndIII 3ul
之后37℃烘箱,不超过16h
十、加5ul lodding buffer
若有条带胶回收。
十一胶回收(步骤同四)
十二做连接,连接表达载体
目的基因(胶回收的)4ul
载体(1300)1ul
DD水2ul
Buffer 2ul
T4连接酶1ul
过夜16℃,水浴连接。
十三、连接后转化,同“六”
十四、挑条单菌落
跑菌落PCR,若有条带,将剩下的9ul +LB液体+K 加入L管过夜培养。
十五、取L管500ul与500ul 甘油加入EP管中,并与-70℃保存
将L管剩下的菌液取1ul跑菌落PCR(10ul体系)
跑电泳检测结果。
十六提质粒
十七酶切检测
DD水7ul
K-B 1ul
质粒1ul
BamI 0.5ul
HinIII 0.5ul
37℃2.5h后可以检测。