基因外显子与内含子的查找方法
编码区和非编码区和内含子外显子

编码区和非编码区和内含子外显子我们都知道不论真核与原核生物都离不开基因,它储存着生长、发育、凋亡等几乎全部生命过程的信息。
那么基因有着哪些结构呢,接下来从三个层面来讨论基因的构成:一、DNA编码区Coding region基因在结构上,分为编码区和非编码区两部分。
真核生物的编码区是不连续的,分为外显子和内含子,在转录过程中会修剪内含子,并拼合外显子来形成转录产物。
在原核生物中,基因是连续的,也就是说无外显子和内含子之分。
外显子Exon外显子是在preRNA 经过剪切或修饰后,被保留的DNA部分,并最终出现在成熟RNA的基因序列中。
内含子Intron在真核生物中,内含子作为阻断基因的线性表达的一段DNA序列,是在preRNA 经过剪切或修饰后,被切除的DNA序列非编码区Non-coding region非编码区在对基因的表达调控中发挥重要作用,如启动子,增强子,终止子等都位于该区域,有意思的是在人类基因中非编码区的占比超过90%。
它们中的一部分可以转录为功能性RNA,比如tRNA(transfer RNA), rRNA(ribosomal RNA)等;可以作为DNA复制,转录起始来对复制,转录和翻译起到调控作用;也可能是着丝粒与端粒的重要组成部分。
启动子Promoter启动子是特定基因转录的DNA区域,启动子一般位于基因的转录起始位点,5‘端上游,启动子长约100-1000bp。
在转录过程中,RNA聚合酶与转录因子可以识别并特异性结合到启动子特有的DNA序列(一般为保守序列),从而启动转录。
启动子本身并不转录而且也不控制基因活动,而是通过转录因子结合来调控转录过程。
在细胞核中,似乎启动子优先分布在染色体区域的边缘,可能是在不同染色体上共同表达基因。
此外,在人类中,启动子显示出每个染色体特有的某些结构特征。
CAAT Box 与Sextama boxCCAAT box(有时也缩写为CAAT box或CAT box):具有GGCCAATCT 共有序列的不同核苷酸序列,是真核生物基因常有的调节区,位于转录起始点上游约-80bp处,可能也是RNA聚合酶的一个结合处,控制着转录起始的频率。
应用UCSC_Ensembl查找基因启动子(promoter)、内含子、外显子序列-表观遗传学论坛-生物秀论坛
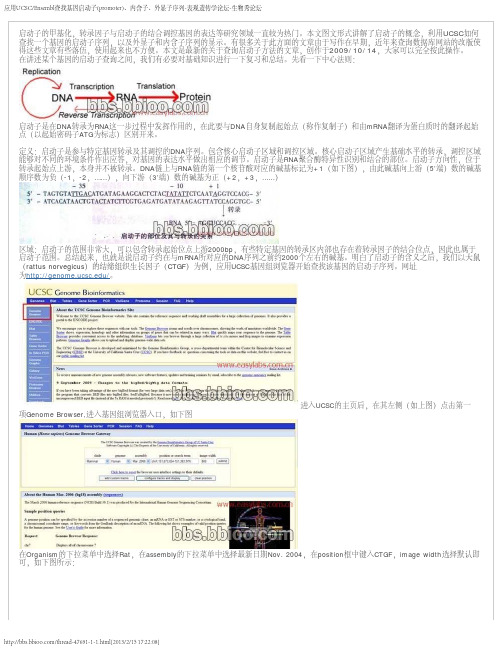
在讲述某个基因的启动子查询之间,我们有必要对基础知识进行一下复习和总结。
先看一下中心法则:启动子是在DNA转录为RNA这一步过程中发挥作用的,在此要与顺序数为负(-1,-2,……),向下游(3’端)数的碱基为正(+2,+3,……)区域:启动子的范围非常大,可以包含转录起始位点上游2000bp,有些特定基因的转录区内部也存在着转录因子的结合位点,因此也属于为/。
进入项Genome Browser,进入基因组浏览器入口,如下图在Organism的下拉菜单中选择Rat,在assembly的下拉菜单中选择最新日期Nov. 2004可,如下图所示:然后点击Submit,返回的页面如下:结果显示该基因的已知序列和相关mRNA序列,点击Known Gene中的第一个序列,出现包含这序列的图解概要。
为了获得这个区域更清晰的图像,可以点击紧靠zoom out的1.5X按钮,如下图:对于Known Genes(已知基因)和预测的基因路径来说,一般的惯例是以一个高的垂直线或块状表示每个编码外显子,以短的垂直线或块状表示5′端和3′端非翻译区。
起连接作用的内含子以非常细的线条表示。
翻译的方向由沿着细线的箭头指示。
本例的搜寻目的来说,默认设置不是理想的设置。
按照视图利用页面底部的Track Controls按钮,将一些路径设置为hide模式(即不显示),其他设置为dense模式(所有资料密集在一条直线上);另一些路径设置为full模式(每个特征有一个分开的线条,最多达300)。
在考虑这些路径内究竟存在那些资料之前,对这些路径的内容和表现做一个简要的讨论是必要的,许多这些讨论是由外界提供给UCSC的。
Ensembl Gene Predictions路径由Ensembl提供。
Ensembl基因通过许多方法来预测,包括与已知mRNA和蛋白质进行同源性比较。
若查询启动子区域,我们需要将Ensembl Genes选择为dense 或full模式,点击Refresh,即刷新,出现下图:图中多出了Ensembl Genes的预测路径,我们在红框中圈出。
内含子和外显子

我把我的理解和大家讨论,理解错误的地方请大家指正首先要明确不是所有的基因都有内含子和外显子组成。
所谓内含子和外显子指的是一个开放阅读框(ORF)内编码的部分和不编码的部分。
内含子和外显子是间隔排列的,所以真核生物这些具有内含子和外显子的基因(不是所有的真核基因都这样)又叫做镶嵌基因或者断裂基因,更早的概念把具有多个内含子外显子的基因叫做多顺反子。
(很抱歉我当时把这个概念弄错了,多顺反子应该指的是一个转录本中共含有多个ORF,在此像战友们道歉。
)我们所说的UTR,一般值得是一个转录本(transcript)3…和5‟不参与编码的区域(但是不是说他们没有功能,只是说他们不被翻译成功能的蛋白质等等)。
我还没有看到用UTR来指代内含子区域的资料,因此UTR 既不指代intron nor Exon。
如果非要说他属于什么,我认为他属于“non-coding region”。
(这个我也错了,wilipedia 解释的很清楚了,希望大家没有被我的错误误导。
但是我仍然是有疑问的,wikipedia的原话是“Some of the exons will be wholly or part of the 5' untranslated region (5' UTR) or the 3' untranslated region (3' UTR) of each transcript.”,为什么是外显子会“部分的”作为UTR?所以我认为UTR不等同于exon。
)明白了问题1,就应该知道promoter自然不属于intron和Exon的任何一个,他就是属于noncoding sequence。
基因间的序列是基因间的序列,和intron没有关系,概念上可以看的出来。
我也不知道到底应该叫他什么。
但是这些序列相当一部分是有功能的。
下举例说明。
大家都知道,noncoding RNA是现在研究的热点之一。
外显子、内含子、mRNA、CDS、ORF区别详解

外显子、内含子、m RNA、CDS、ORF区别与联系1、DN A复制:以DN A为模板,在DN A聚合酶的催化作用下,将四种游离的d NTP按照碱基互补配对原则合成新链DNA转录:以DN A为模版,在DN A指导的RNA聚合酶的作用下,将四种游离的NTP按照碱基互补配对的原则合成RNA翻译:以m RNA为模板,在核糖体内合成蛋白质的过程特点:DN A复制:模板为双链DN A,合成的新链与模板链一模一样,原料为四种d NTP,为半保留复制,需要引物转录:模板为双链DN A,为半不连续转录需要引物,原料为四种NTP,合成的新链除了把DN A上的T改为U外,其他一样翻译:模板为m RNA,原料为20中游离的氨基酸,3个碱基决定一个氨基酸2、m RNAm RNA(m e sse n g e r RNA,信使RNA)信使RNA是由DN A经h n RNA剪接而成,携带遗传信息的能指导蛋白合成的一类单链核糖核酸。
3、基因DN A分为编码区和非编码区,编码区包含外显子和内含子,一般非编码区具有基因表达的调控功能,如启动子在非编码区。
编码区则转录为m RNA 并最终翻译成蛋白质。
外显子和内含子都被转录到m RNA前体hnRNA中,当hnRNA进行剪接变为成熟的m RNA时,内含子被切除,而外显子保留。
实际上真正编码蛋白质的是外显子,而内含子则无编码功能,内含子存在于DN A中,在转录的过程中,DN A上的内含子也会被转录到前体RNA中,但前体RNA上的内含子会在RNA离开细胞核进行翻译前被切除。
4、CDS Se q u e n ceco d in gfo r am in oacid s inp ro t e in蛋白质编码区CDS是Co dingse que nce的缩写,是编码一段蛋白产物的序列,是结构基因组学术语。
与开放读码框ORF的区别开放读码框是从一个起始密码子开始到一个终止密码子结束的一段序列;不是所有读码框都能被表达出蛋白产物,或者能表达出占有优势或者能产生生物学功能的蛋白。
内含子和外显子的位置

偶最近也在做RT-PCR。
也正在设计跨内含子的引物。
在园子里瞧了些文献,也请教了一些人。
个人感觉主要就是您找到这个基因的具体的内含子与外显子的位置后再设计引物一点也不难。
只要在跨越的引物中选取条件好的就行了。
其中对于如何找到内含子与外显子,偶就是用以下的方法。
一、首先登录Pubmed找到您的基因序列,这个应该很简单。
偶不多说。
二、如下图进入Blastl界面,选取genomes中的相应物种。
三将您的序列拷贝到查询框中,进行查询(Begin search)。
然后点击format,一路点下去,出现下图样的对话框。
点击genome view然后点击其相应的染色体号,我查的就是HIF基因,在14号染色体上。
相应的染色体上会出现红色的标记。
好了,如下图。
您可以瞧到右边的这些方框最后给的序列,就就是外显子的序列。
您可以对着此来设计引物,找到哪些跨过内含子的引物。
首先要明白:RT-PCR的引物之所以要跨内含子设计,主要原因就就是为了避免再PCR过程中基因组DNA的影响。
那么其实只要在提RNA时除去DNA也就没什么关系了。
RNA沉淀之后,在100ul反应体系中用RNAse free的DNAseI37C处理0、5h,然后直接用100ul氯仿抽提,异丙醇(别忘了加1/10体积4M NaAc)再次沉淀后用适量DEPC水溶解即可。
NCBI>GENE>基因名称搜索(Click the picture to see the source) louischenPosts:518Score:432004-07-30 06:57点击基因名称进入左键击NC_000019>GRAPHICSintron,extron的排列,可以点击进入粗划线为外显子,并有对应的protein编码还在也可以,在主页上输入基因的名字,找出该物种的基因序列,在transview与exonview上可以瞧到外显子与内含子,此网页简单,得到不同颜色标记的外显子。
NCBI查基因 编码区 启动子区 外显子
问题:NCBI中怎样查找编码区/非编码区、起始密码子、启动子、外显子/内含子。
启动子
一般定义启动子,都是upstream 1000bp,downstream1000bp的那段序列。
或者根据你的实验。
你去ensemble,输入基因,找到exon,点开,在configuration里面选好flank多少bp 的序列,选好之后自动刷新,就出来了。
在序列里面,ensemble用不同的颜色标出来不同区域,5‘UTR之类的,还有exon,intron,转录起始位点等等,flank的区域就是你选的promoter 了。
开放阅读框
在分子生物学中,开放阅读框(Open Reading Frame, ORF)从起始密码子开始,是DNA序列中具有编码蛋白质潜能,一段无终止密码子打断的碱基序列。
基因外显子与内含子的查找方法

基因外显子与内含子的查找方法一、外显子/内含子的概念1. 外显子(exon) sequence of a gene's DNA that transcribes into protein structures外显子(expressed region) 是真核生物基因的一部分,它在剪接(Splicing)后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质。
外显子是最后出现在成熟RNA中的基因序列, 又称表达序列。
既存在于最初的转录产物中,也存在于成熟的RNA分子中的核苷酸序列。
术语外显子也指编码相应RNA外显子的DNA中的区域。
*简言之,外显子就是指真核细胞的基因在表达过程中能编码蛋白质的核苷酸序列。
☆重要特点:①比较不同物种的相关基因,我们发现相应的外显子序列通常是保守的,而内含子序列则很少保守。
②编码蛋白质的序列通常处于选择压力之下,内含子由于没有选择压力,因此比外显子的进化快得多。
③通过确定在多种生物中出现的片段来鉴定编码区域,而外显子的保守性可以作为这种鉴定的基础2. 内含子(introns)内含子是基因内的间隔序列,不出现在成熟的RNA分子中,在转录后通过加工被切除。
大多数真核生物的基因都有内含子。
需注意的是,在古细菌中也有内含子。
在转录后的加工中,从最初的转录产物除去的内部的核苷酸序列。
术语内含子也指编码相应RNA内含子的DNA中的区域。
大多数真核结构基因中的间插序列(intervening sequence)或不编码序列。
它们可以转录,但在基因转录后,由这些间插序列转录的部分(也可用内含子这个术语表示)经加工被从初级转录本中准确除去,才产生有功能的RNA。
基因的编码部分称外显子。
内含子常比外显子长,且占基因的更大比例。
真核基因所含内含子的数目、位置和长度不尽相同,如鸡卵清蛋白基因的外显子被7个内含子隔开,鸡卵伴清蛋白基因有17个内含子,α-珠蛋白基因有2个内含子,卵粘蛋白基因有6个内含子等。
外显子、内含子、mRNA、CDS、ORF区别与联系

外显子、内含子、mRNA、CDS、ORF区别与联系1、DNA复制:以DNA为模板,在DNA聚合酶的催化作用下,将四种游离的dNTP按照碱基互补配对原则合成新链DNA转录:以DNA为模版,在DNA指导的RNA聚合酶的作用下,将四种游离的NTP按照碱基互补配对的原则合成RNA翻译:以mRNA为模板,在核糖体内合成蛋白质的过程特点:DNA复制?:? 模板为双链DNA,合成的新链与模板链一模一样,原料为四种dNTP,为半保留复制,需要引物转录:模板为双链DNA,为半不连续转录需要引物,原料为四种NTP,合成的新链除了把DNA上的T改为U外,其他一样?翻译?:模板为mRNA,原料为20中游离的氨基酸,3个碱基决定一个氨基酸2、mRNAmRNA (messenger RNA,信使RNA)信使RNA是由DNA经hnRNA剪接而成,携带遗传信息的能指导蛋白合成的一类单链核糖核酸。
3、基因DNA分为编码区和非编码区,编码区包含外显子和内含子,一般非编码区具有基因表达的调控功能,如启动子在非编码区。
编码区则转录为mRNA 并最终翻译成蛋白质。
外显子和内含子都被转录到mRNA前体hnRNA中,当hnRNA进行剪接变为成熟的mRNA时,内含子被切除,而外显子保留。
实际上真正编码蛋白质的是外显子,而内含子则无编码功能,内含子存在于DNA中,在转录的过程中,DNA上的内含子也会被转录到前体RNA中,但前体RNA上的内含子会在RNA离开细胞核进行翻译前被切除。
4、CDS? Sequence coding for amino acids in protein 蛋白质编码区?? ? CDS是Coding sequence的缩写,是编码一段蛋白产物的序列,是结构基因组学术语。
与开放读码框ORF的区别开放读码框是从一个起始密码子开始到一个终止密码子结束的一段序列;不是所有读码框都能被表达出蛋白产物,或者能表达出占有优势或者能产生生物学功能的蛋白。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
基因外显子与内含子的查找方法
基因的内含子和外显子的序列
在NCBI找到基因序列之后,要确定其内含子和外显子的序列是比较简单的。
如果是RefSeq 序列的话,那就更加容易了。
简单的思路就是跟基因组序列比对就行了。
% 用NCBI的Splign工具,详见:图解:如何在NCBI上找到HNF-4基因第4个外显子的序列。
% 但由于猪KIT(FJ938289)基因不是Refseq序列。
这就比较难了,由该序列的注释可知(/chromosome="8),该序列是属于第8号染色体。
% 所以先确定猪(Pig,Sus scrofa)的第8号染色体是否已经测完序了。
先在Taxonomy 数据库搜索,得到Taxonomy ID: 9823。
直接用关键词txid9823[Organism:noexp] 搜索Genome数据库。
猪有19条染色体,结果为11。
看图:
% 但其中并没有第8号染色体的。
所以要确定猪KIT(FJ938289)基因的内含子和外显子的序列,好像就不大可能了。
反正我是没办法了。
基因结构图
1,NCBI提供的Graphics工具
在NCBI搜索FJ938289之后,接下来用NCBI提供的Graphics工具来查看,就能知道基因的内含子和外显子的序列了(同时也是一个基因结构图哦)。
看图:。